Department of Molecular and Cell Biology, California Institute of Quantitative Biosciences, University of California Berkeley, Berkeley, California, United States of America.
PLoS Genet. 2011 Feb 3;7(2):e1001290. doi: 10.1371/journal.pgen.1001290.
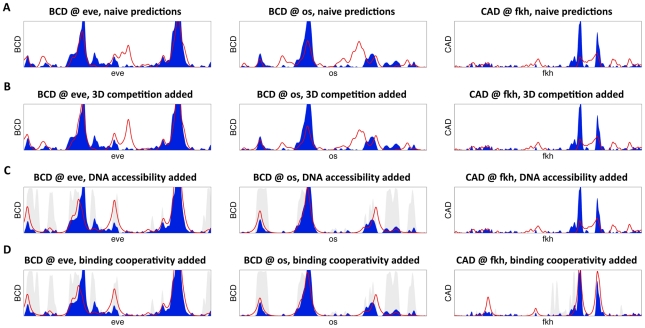
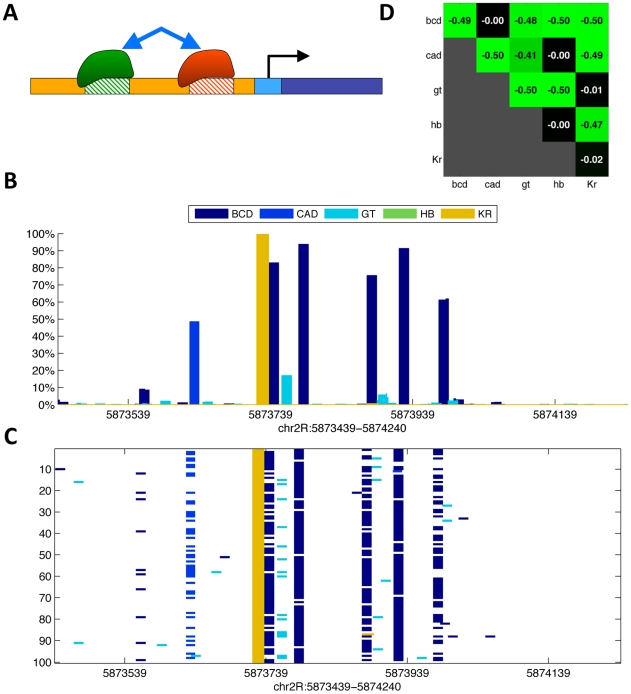
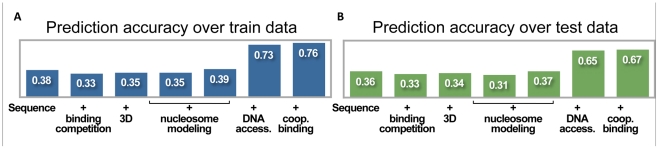
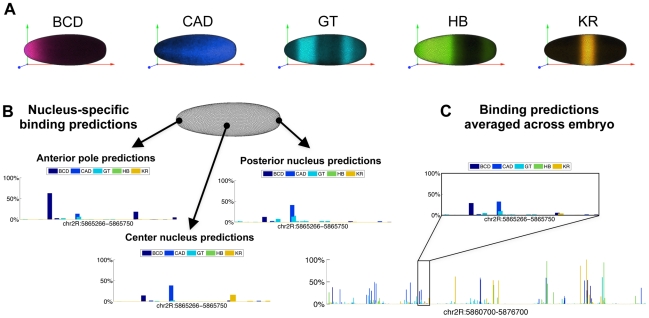
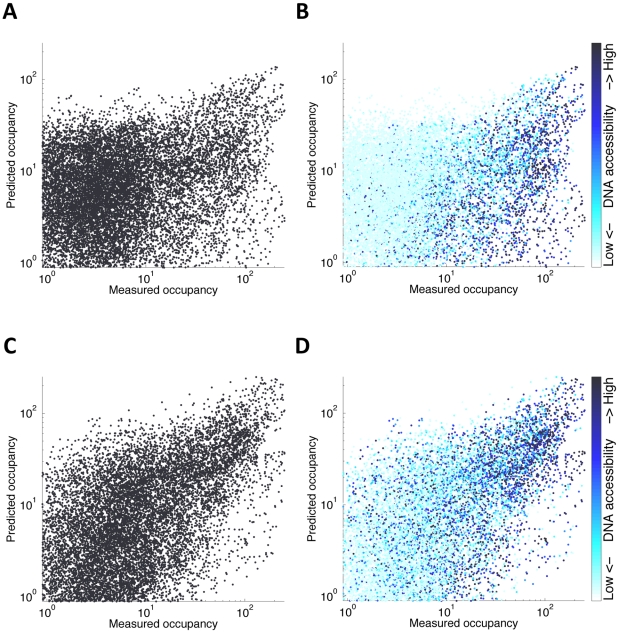
Transcription factors that drive complex patterns of gene expression during animal development bind to thousands of genomic regions, with quantitative differences in binding across bound regions mediating their activity. While we now have tools to characterize the DNA affinities of these proteins and to precisely measure their genome-wide distribution in vivo, our understanding of the forces that determine where, when, and to what extent they bind remains primitive. Here we use a thermodynamic model of transcription factor binding to evaluate the contribution of different biophysical forces to the binding of five regulators of early embryonic anterior-posterior patterning in Drosophila melanogaster. Predictions based on DNA sequence and in vitro protein-DNA affinities alone achieve a correlation of ∼0.4 with experimental measurements of in vivo binding. Incorporating cooperativity and competition among the five factors, and accounting for spatial patterning by modeling binding in every nucleus independently, had little effect on prediction accuracy. A major source of error was the prediction of binding events that do not occur in vivo, which we hypothesized reflected reduced accessibility of chromatin. To test this, we incorporated experimental measurements of genome-wide DNA accessibility into our model, effectively restricting predicted binding to regions of open chromatin. This dramatically improved our predictions to a correlation of 0.6-0.9 for various factors across known target genes. Finally, we used our model to quantify the roles of DNA sequence, accessibility, and binding competition and cooperativity. Our results show that, in regions of open chromatin, binding can be predicted almost exclusively by the sequence specificity of individual factors, with a minimal role for protein interactions. We suggest that a combination of experimentally determined chromatin accessibility data and simple computational models of transcription factor binding may be used to predict the binding landscape of any animal transcription factor with significant precision.
在动物发育过程中,驱动复杂基因表达模式的转录因子与数千个基因组区域结合,结合区域的结合定量差异调节其活性。虽然我们现在有工具来描述这些蛋白质的 DNA 亲和力,并精确测量它们在体内的全基因组分布,但我们对决定它们结合的位置、时间和程度的力量的理解仍然很原始。在这里,我们使用转录因子结合的热力学模型来评估不同生物物理力对五种调节果蝇早期胚胎前后模式形成的转录因子在体内结合的贡献。仅基于 DNA 序列和体外蛋白质-DNA 亲和力的预测与体内结合的实验测量相关性约为 0.4。纳入五个因素之间的协同作用和竞争,并通过独立建模每个核中的结合来解释空间模式,对预测准确性几乎没有影响。一个主要的误差源是预测体内不存在的结合事件,我们假设这反映了染色质的可及性降低。为了验证这一点,我们将全基因组 DNA 可及性的实验测量值纳入我们的模型中,有效地将预测的结合限制在开放染色质区域。这极大地提高了我们的预测准确性,对于各种因素在已知靶基因上的相关性达到 0.6-0.9。最后,我们使用我们的模型来量化 DNA 序列、可及性和结合竞争与协同作用的作用。我们的结果表明,在开放染色质区域,结合可以几乎完全由单个因素的序列特异性来预测,蛋白质相互作用的作用很小。我们建议,可以结合实验确定的染色质可及性数据和转录因子结合的简单计算模型,以相当高的精度预测任何动物转录因子的结合景观。