Department of Molecular Biology and Genetics, Cornell University, Ithaca, New York, United States of America.
PLoS Genet. 2012;8(3):e1002610. doi: 10.1371/journal.pgen.1002610. Epub 2012 Mar 29.
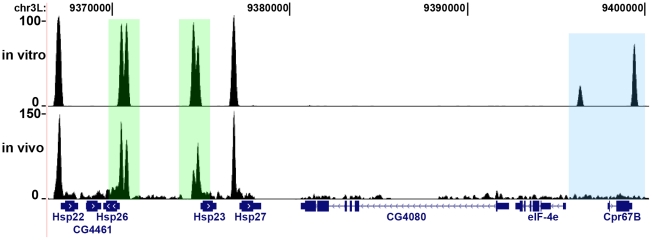
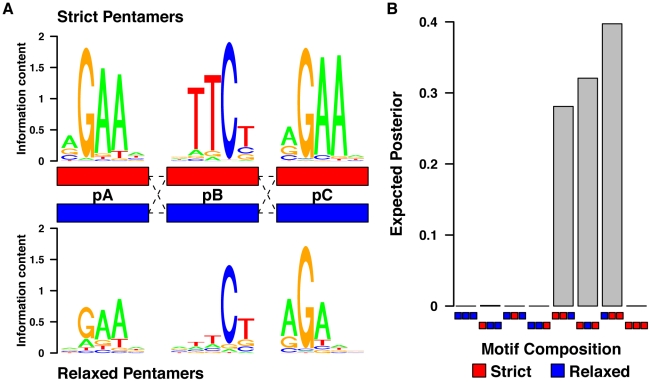
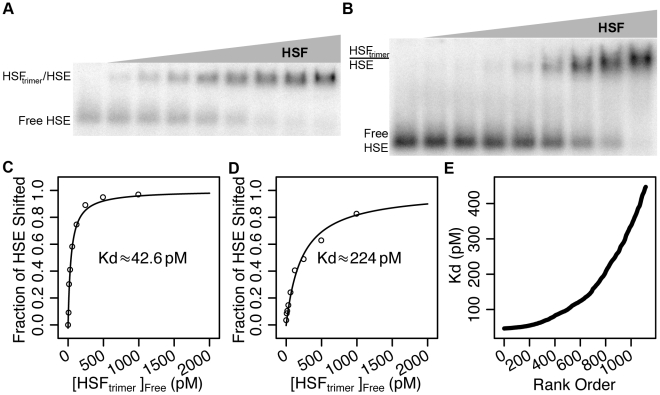
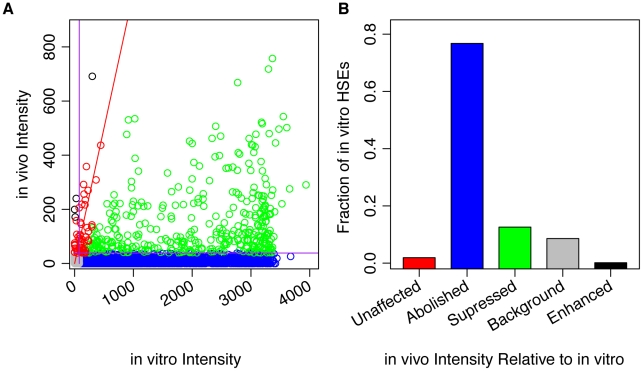
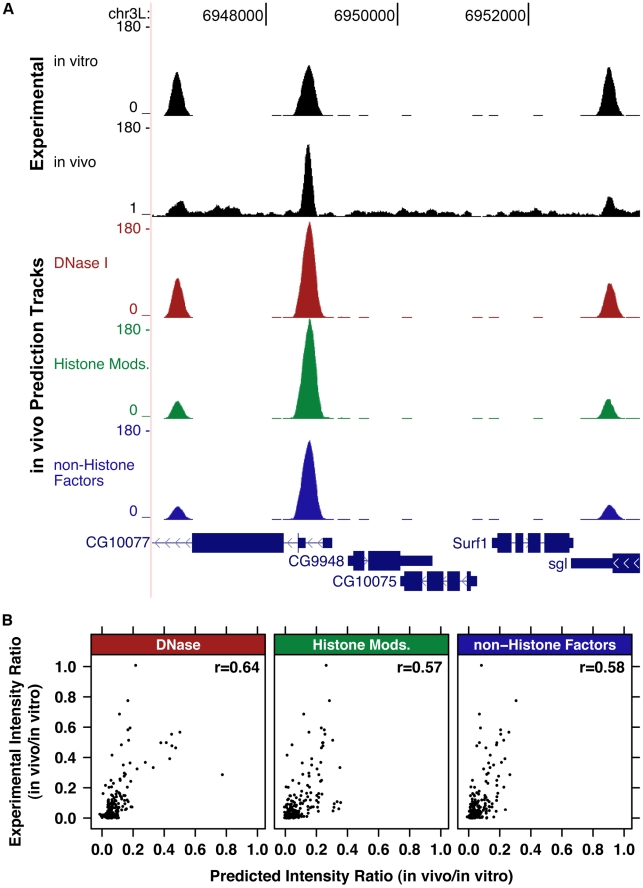
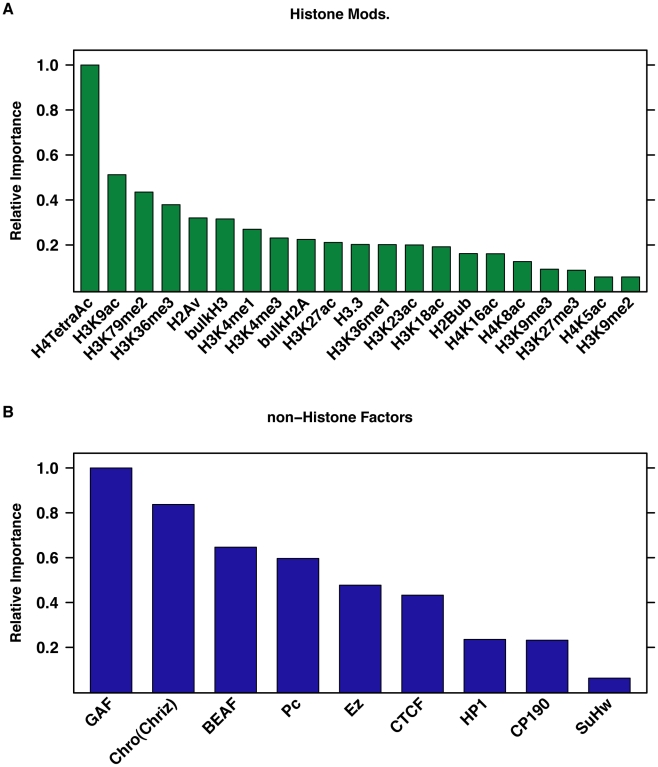
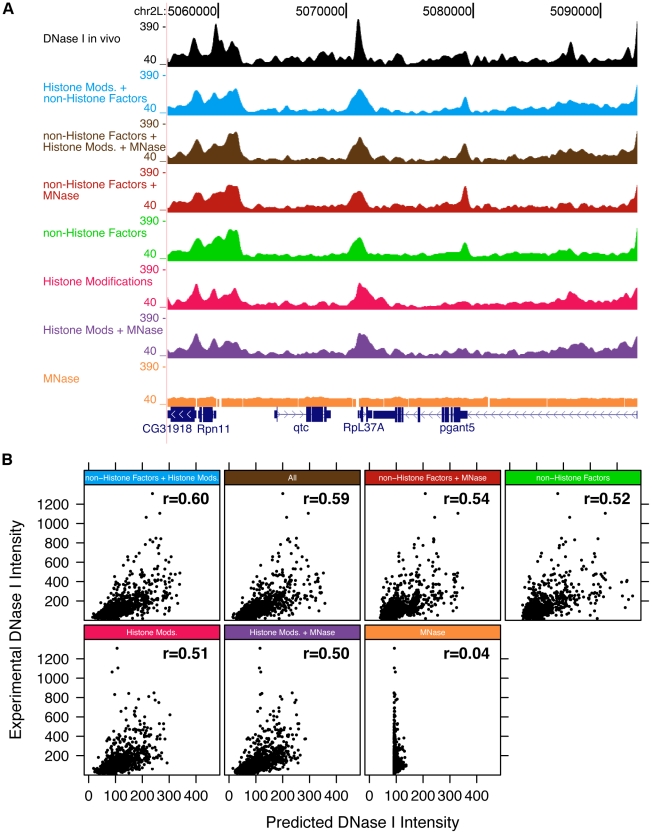
DNA sequence and local chromatin landscape act jointly to determine transcription factor (TF) binding intensity profiles. To disentangle these influences, we developed an experimental approach, called protein/DNA binding followed by high-throughput sequencing (PB-seq), that allows the binding energy landscape to be characterized genome-wide in the absence of chromatin. We applied our methods to the Drosophila Heat Shock Factor (HSF), which inducibly binds a target DNA sequence element (HSE) following heat shock stress. PB-seq involves incubating sheared naked genomic DNA with recombinant HSF, partitioning the HSF-bound and HSF-free DNA, and then detecting HSF-bound DNA by high-throughput sequencing. We compared PB-seq binding profiles with ones observed in vivo by ChIP-seq and developed statistical models to predict the observed departures from idealized binding patterns based on covariates describing the local chromatin environment. We found that DNase I hypersensitivity and tetra-acetylation of H4 were the most influential covariates in predicting changes in HSF binding affinity. We also investigated the extent to which DNA accessibility, as measured by digital DNase I footprinting data, could be predicted from MNase-seq data and the ChIP-chip profiles for many histone modifications and TFs, and found GAGA element associated factor (GAF), tetra-acetylation of H4, and H4K16 acetylation to be the most predictive covariates. Lastly, we generated an unbiased model of HSF binding sequences, which revealed distinct biophysical properties of the HSF/HSE interaction and a previously unrecognized substructure within the HSE. These findings provide new insights into the interplay between the genomic sequence and the chromatin landscape in determining transcription factor binding intensity.
DNA 序列和局部染色质结构共同决定了转录因子(TF)的结合强度谱。为了理清这些影响,我们开发了一种实验方法,称为蛋白/DNA 结合后高通量测序(PB-seq),该方法允许在没有染色质的情况下,对全基因组的结合能谱进行特征描述。我们将该方法应用于果蝇热休克因子(HSF),该因子在受到热休克应激后可诱导地结合靶 DNA 序列元件(HSE)。PB-seq 涉及用重组 HSF 孵育剪切的裸露基因组 DNA,将 HSF 结合和 HSF 游离的 DNA 分开,然后通过高通量测序检测 HSF 结合的 DNA。我们将 PB-seq 结合谱与体内 ChIP-seq 观察到的结合谱进行了比较,并开发了统计模型,根据描述局部染色质环境的协变量来预测观察到的与理想化结合模式的偏差。我们发现,DNase I 超敏性和 H4 的四乙酰化是预测 HSF 结合亲和力变化的最具影响力的协变量。我们还研究了 DNA 可及性(通过数字 DNase I 足迹数据测量)在多大程度上可以从 MNase-seq 数据和许多组蛋白修饰和 TF 的 ChIP-chip 图谱中预测出来,发现 GAGA 元件相关因子(GAF)、H4 的四乙酰化和 H4K16 乙酰化是最具预测性的协变量。最后,我们生成了一个 HSF 结合序列的无偏模型,该模型揭示了 HSF/HSE 相互作用的独特物理性质和 HSE 内的一个以前未被识别的亚结构。这些发现为确定转录因子结合强度的基因组序列和染色质景观之间的相互作用提供了新的见解。