School of Computer Science and Engineering, Seoul National University, Seoul 151-742, Korea.
BMC Bioinformatics. 2011 Feb 15;12 Suppl 1(Suppl 1):S46. doi: 10.1186/1471-2105-12-S1-S46.
Protein quantification is an essential step in many proteomics experiments. A number of labeling approaches have been proposed and adopted in mass spectrometry (MS) based relative quantification. The mTRAQ, one of the stable isotope labeling methods, is amine-specific and available in triplex format, so that the sample throughput could be doubled when compared with duplex reagents.
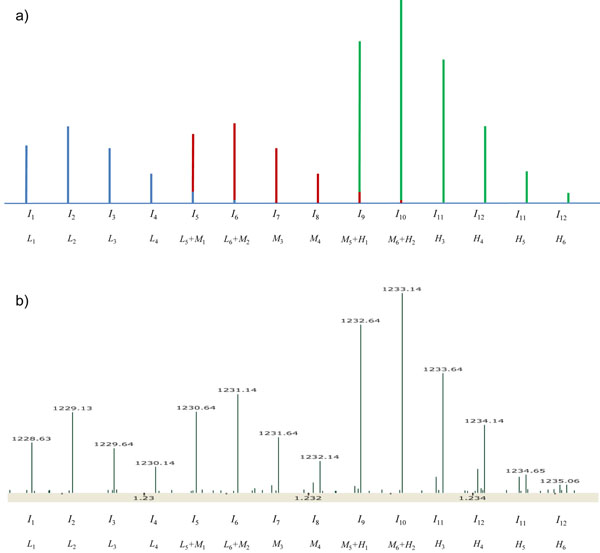
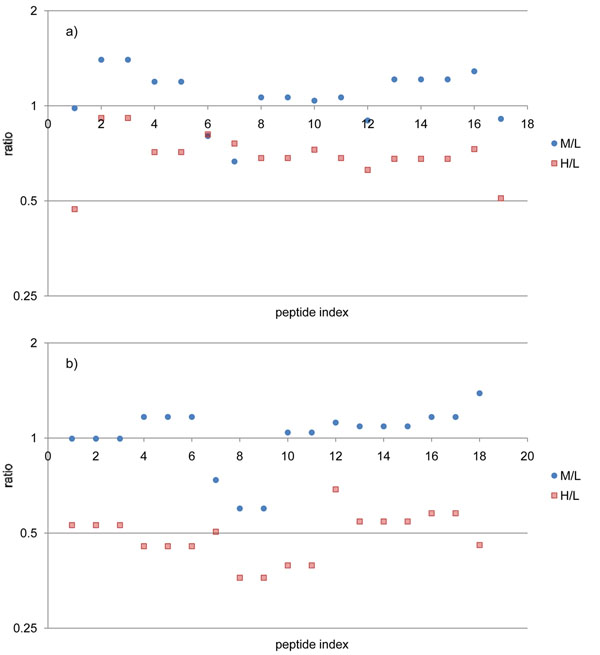
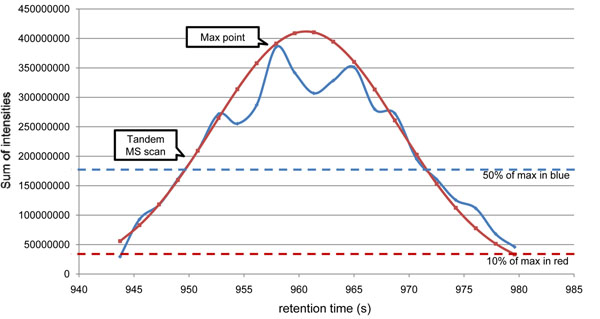
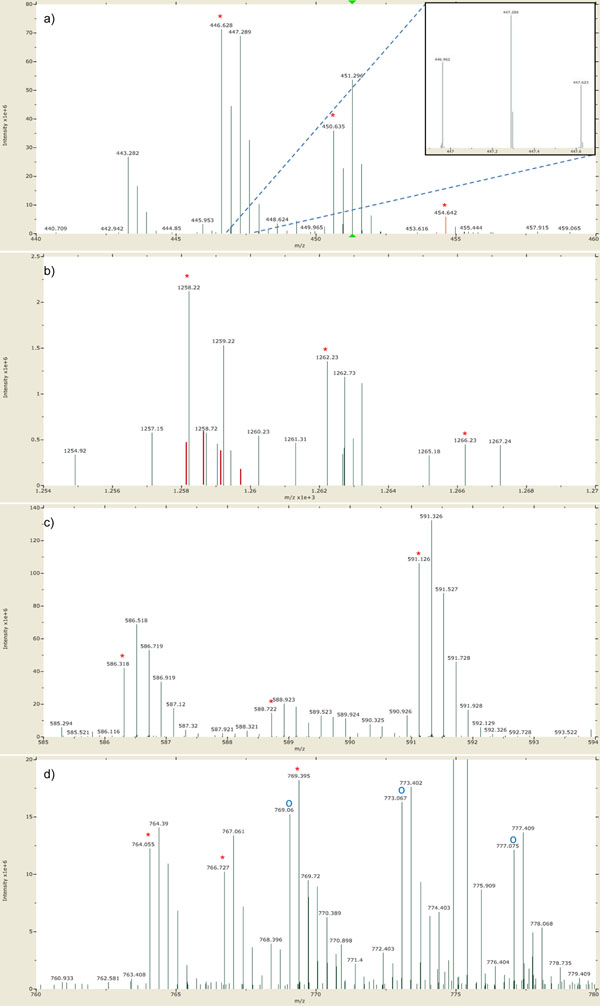
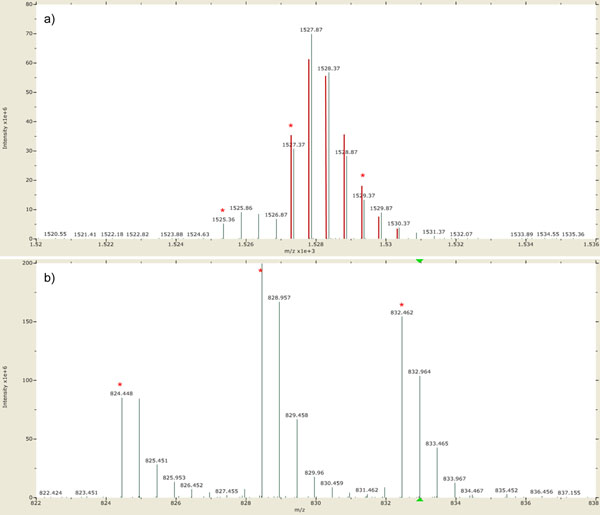
Here we propose a novel data analysis algorithm for peptide quantification in triplex mTRAQ experiments. It improved the accuracy of quantification in two features. First, it identified and separated triplex isotopic clusters of a peptide in each full MS scan. We designed a schematic model of triplex overlapping isotopic clusters, and separated triplex isotopic clusters by solving cubic equations, which are deduced from the schematic model. Second, it automatically determined the elution areas of peptides. Some peptides have similar atomic masses and elution times, so their elution areas can have overlaps. Our algorithm successfully identified the overlaps and found accurate elution areas. We validated our algorithm using standard protein mixture experiments.
We showed that our algorithm was able to accurately quantify peptides in triplex mTRAQ experiments. Its software implementation is compatible with Trans-Proteomic Pipeline (TPP), and thus enables high-throughput analysis of proteomics data.
蛋白质定量是许多蛋白质组学实验的重要步骤。在基于质谱(MS)的相对定量中,已经提出并采用了许多标记方法。mTRAQ 是一种稳定同位素标记方法,具有胺特异性,可采用三联体格式,与双试剂相比,可将样品通量提高一倍。
在这里,我们提出了一种用于三联体 mTRAQ 实验中肽定量的新数据分析算法。它通过两个方面提高了定量的准确性。首先,它在每个全 MS 扫描中鉴定并分离了肽的三联体同位素簇。我们设计了三联体重叠同位素簇的示意模型,并通过从该示意模型推导出的三次方程来分离三联体同位素簇。其次,它自动确定肽的洗脱区域。一些肽具有相似的原子质量和洗脱时间,因此它们的洗脱区域可能会重叠。我们的算法成功地识别了重叠并找到了准确的洗脱区域。我们使用标准蛋白质混合物实验验证了我们的算法。
我们表明,我们的算法能够准确地定量三联体 mTRAQ 实验中的肽。它的软件实现与 Trans-Proteomic Pipeline(TPP)兼容,从而能够实现蛋白质组学数据的高通量分析。