Institute of Computing Technology, Chinese Academy of Sciences, Beijing, China.
BMC Bioinformatics. 2011 Feb 15;12 Suppl 1(Suppl 1):S54. doi: 10.1186/1471-2105-12-S1-S54.
Native structures of proteins are formed essentially due to the combining effects of local and distant (in the sense of sequence) interactions among residues. These interaction information are, explicitly or implicitly, encoded into the scoring function in protein structure prediction approaches--threading approaches usually measure an alignment in the sense that how well a sequence adopts an existing structure; while the energy functions in Ab Initio methods are designed to measure how likely a conformation is near-native. Encouraging progress has been observed in structure refinement where knowledge-based or physics-based potentials are designed to capture distant interactions. Thus, it is interesting to investigate whether distant interaction information captured by the Ab Initio energy function can be used to improve threading, especially for the weakly/distant homologous templates.
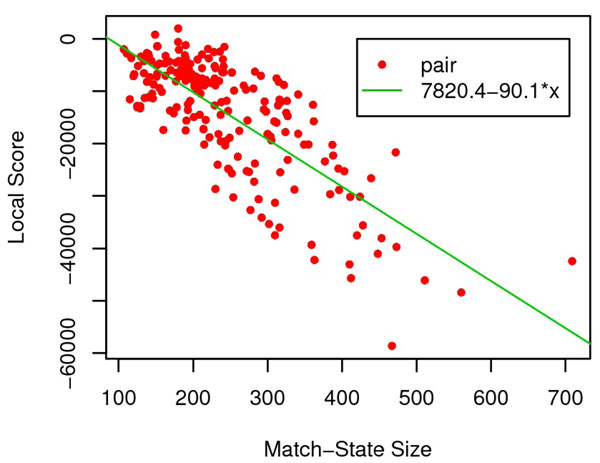
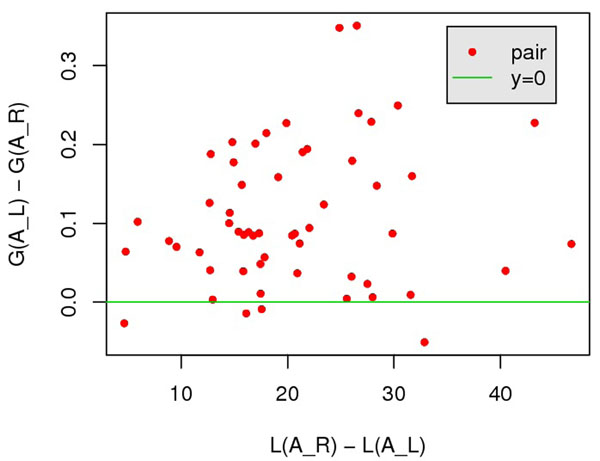
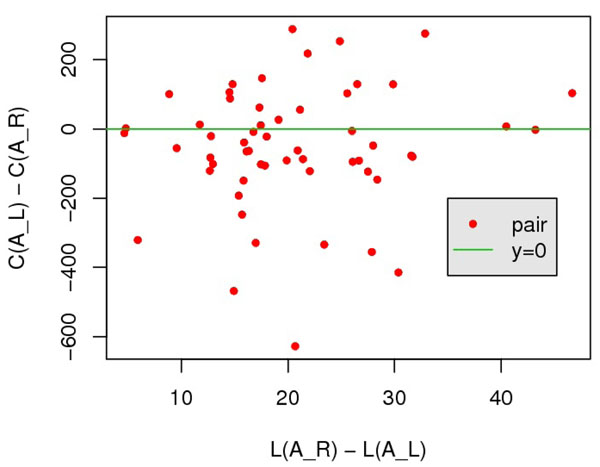
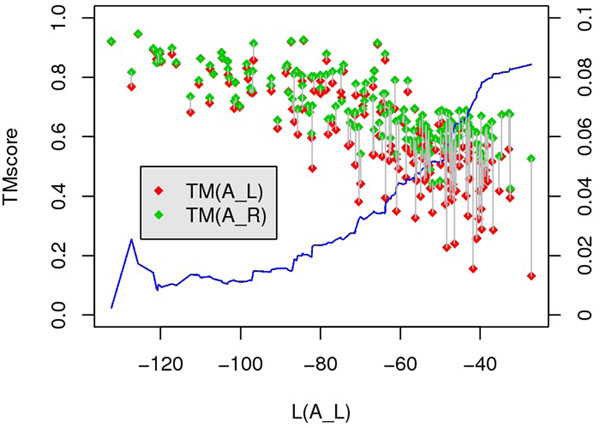
In this paper, we investigate the possibility to improve alignment-generating through incorporating distant interaction information into the alignment scoring function in a nontrivial approach. Specifically, the distant interaction information is introduced through employing an Ab Initio energy function to evaluate the "partial" decoy built from an alignment. Subsequently, a local search algorithm is utilized to optimize the scoring function.Experimental results demonstrate that with distant interaction items, the quality of generated alignments are improved on 68 out of 127 query-template pairs in Prosup benchmark. In addition, compared with state-to-art threading methods, our method performs better on alignment accuracy comparison.
Incorporating Ab Initio energy functions into threading can greatly improve alignment accuracy.
蛋白质的天然结构主要是由于残基之间的局部和远程(在序列意义上)相互作用的组合效应形成的。这些相互作用信息明确或隐含地编码在蛋白质结构预测方法(如 threading 方法)的评分函数中——threading 方法通常以序列适应现有结构的程度来衡量比对;而从头计算方法中的能量函数旨在衡量构象接近天然的可能性。在结构精修方面取得了令人鼓舞的进展,其中基于知识或基于物理的势能被设计用来捕捉远程相互作用。因此,研究从头计算能量函数中捕获的远程相互作用信息是否可以用于改进 threading,特别是对于弱/远程同源模板,这是很有趣的。
在本文中,我们通过以一种非平凡的方式将远程相互作用信息引入到比对评分函数中,研究了通过将远程相互作用信息引入到比对评分函数中以改进比对生成的可能性。具体来说,通过使用从头计算能量函数来评估来自比对的“部分”诱饵,引入远程相互作用信息。随后,利用局部搜索算法来优化评分函数。实验结果表明,在 Prosup 基准测试的 127 个查询-模板对中有 68 个对,引入远程相互作用项可以提高生成比对的质量。此外,与最先进的 threading 方法相比,我们的方法在比对准确性比较方面表现更好。
将从头计算能量函数纳入 threading 可以大大提高比对准确性。