Institute of Biomedical Informatics, National Yang-Ming University, Taipei, Taiwan.
BMC Bioinformatics. 2011 Feb 15;12 Suppl 1(Suppl 1):S9. doi: 10.1186/1471-2105-12-S1-S9.
Un-MAppable Reads Solution (UMARS) is a user-friendly web service focusing on retrieving valuable information from sequence reads that cannot be mapped back to reference genomes. Recently, next-generation sequencing (NGS) technology has emerged as a powerful tool for generating high-throughput sequencing data and has been applied to many kinds of biological research. In a typical analysis, adaptor-trimmed NGS reads were first mapped back to reference sequences, including genomes or transcripts. However, a fraction of NGS reads failed to be mapped back to the reference sequences. Such un-mappable reads are usually imputed to sequencing errors and discarded without further consideration.
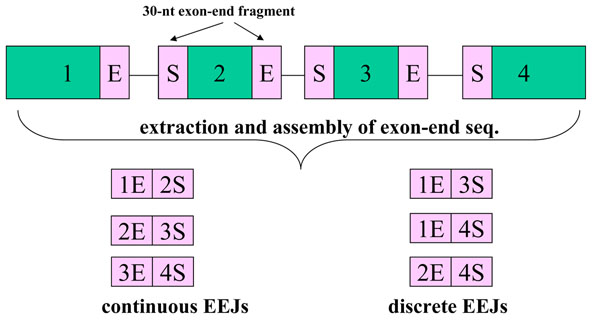
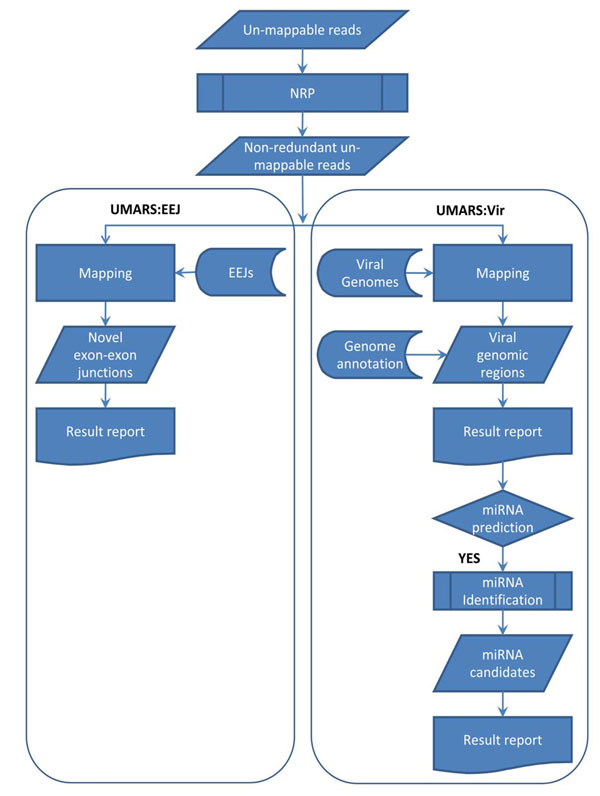
We are investigating possible biological relevance and possible sources of un-mappable reads. Therefore, we developed UMARS to scan for virus genomic fragments or exon-exon junctions of novel alternative splicing isoforms from un-mappable reads. For mapping un-mappable reads, we first collected viral genomes and sequences of exon-exon junctions. Then, we constructed UMARS pipeline as an automatic alignment interface.
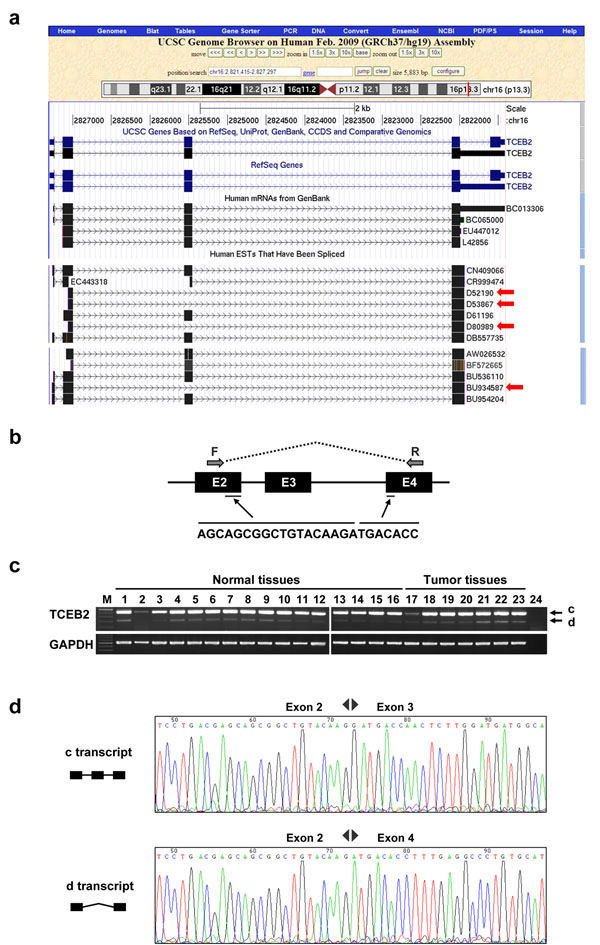
By demonstrating the results of two UMARS alignment cases, we show the applicability of UMARS. We first showed that the expected EBV genomic fragments can be detected by UMARS. Second, we also detected exon-exon junctions from un-mappable reads. Further experimental validation also ensured the authenticity of the UMARS pipeline. The UMARS service is freely available to the academic community and can be accessed via http://musk.ibms.sinica.edu.tw/UMARS/.
In this study, we have shown that some un-mappable reads are not caused by sequencing errors. They can originate from viral infection or transcript splicing. Our UMARS pipeline provides another way to examine and recycle the un-mappable reads that are commonly discarded as garbage.
Un-MAppable Reads Solution(UMARS)是一个用户友好的网络服务,专注于从无法映射回参考基因组的序列读取中检索有价值的信息。最近,下一代测序(NGS)技术已成为生成高通量测序数据的强大工具,并已应用于许多种生物研究。在典型的分析中,首先将接头修剪的 NGS 读取映射回参考序列,包括基因组或转录本。然而,一部分 NGS 读取无法映射回参考序列。这些无法映射的读取通常被归因于测序错误,并在没有进一步考虑的情况下被丢弃。
我们正在研究无法映射的读取可能具有的生物学相关性和可能的来源。因此,我们开发了 UMARS,以从无法映射的读取中扫描病毒基因组片段或新型可变剪接异构体的外显子-外显子连接。为了映射无法映射的读取,我们首先收集病毒基因组和外显子-外显子连接的序列。然后,我们构建了 UMARS 管道作为自动对齐接口。
通过展示两个 UMARS 对齐案例的结果,我们展示了 UMARS 的适用性。我们首先表明,UMARS 可以检测到预期的 EBV 基因组片段。其次,我们还从无法映射的读取中检测到外显子-外显子连接。进一步的实验验证也确保了 UMARS 管道的真实性。UMARS 服务免费提供给学术界,可以通过 http://musk.ibms.sinica.edu.tw/UMARS/ 访问。
在这项研究中,我们表明,一些无法映射的读取不是由测序错误引起的。它们可能源自病毒感染或转录剪接。我们的 UMARS 管道提供了另一种检查和回收通常被视为垃圾的无法映射读取的方法。