Stazione Zoologica Anton Dohrn, Villa Comunale, Napoli, 80121, Italy.
High Performance Computing and Networking Institute, National Research Council of Italy, Via P. Castellino, 111, Napoli, 80131, Italy.
BMC Bioinformatics. 2019 Apr 18;20(Suppl 4):168. doi: 10.1186/s12859-019-2684-x.
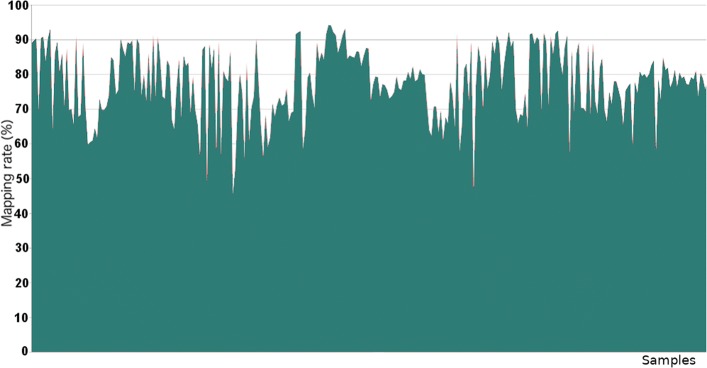
Next Generation Sequencing (NGS) experiments produce millions of short sequences that, mapped to a reference genome, provide biological insights at genomic, transcriptomic and epigenomic level. Typically the amount of reads that correctly maps to the reference genome ranges between 70% and 90%, leaving in some cases a consistent fraction of unmapped sequences. This 'misalignment' can be ascribed to low quality bases or sequence differences between the sample reads and the reference genome. Investigating the source of the unmapped reads is definitely important to better assess the quality of the whole experiment and to check for possible downstream or upstream 'contamination' from exogenous nucleic acids.
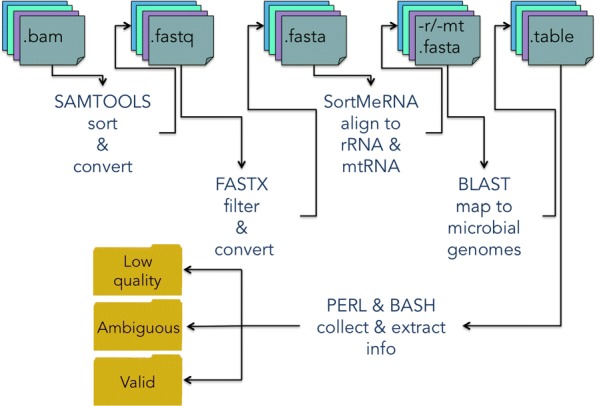
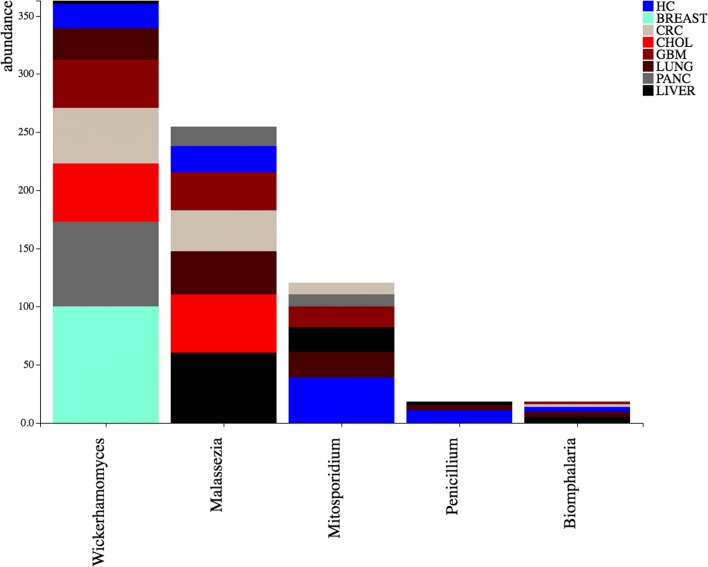
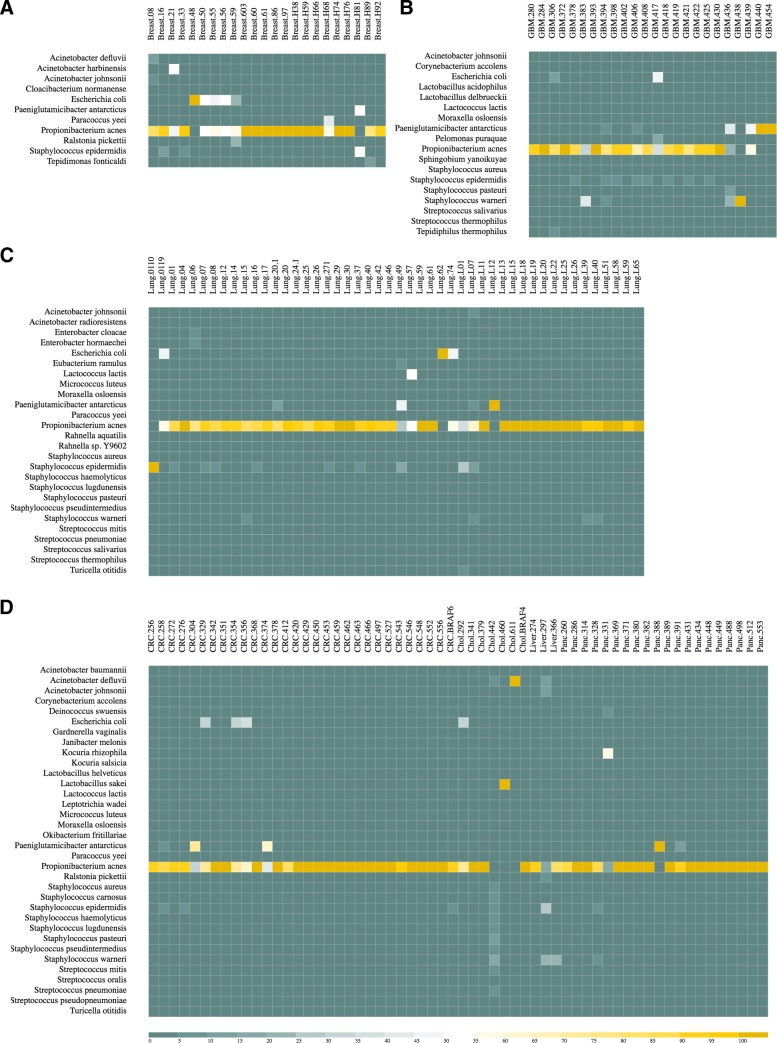
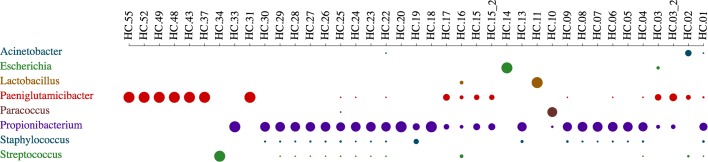
Here we propose DecontaMiner, a tool to unravel the presence of contaminating sequences among the unmapped reads. It uses a subtraction approach to identify bacteria, fungi and viruses genome contamination. DecontaMiner generates several output files to track all the processed reads, and to provide a complete report of their characteristics. The good quality matches on microorganism genomes are counted and compared among samples. DecontaMiner builds an offline HTML page containing summary statistics and plots. The latter are obtained using the state-of-the-art D3 javascript libraries. DecontaMiner has been mainly used to detect contamination in human RNA-Seq data. The software is freely available at http://www-labgtp.na.icar.cnr.it/decontaminer .
DecontaMiner is a tool designed and developed to investigate the presence of contaminating sequences in unmapped NGS data. It can suggest the presence of contaminating organisms in sequenced samples, that might derive either from laboratory contamination or from their biological source, and in both cases can be considered as worthy of further investigation and experimental validation. The novelty of DecontaMiner is mainly represented by its easy integration with the standard procedures of NGS data analysis, while providing a complete, reliable, and automatic pipeline.
下一代测序(NGS)实验产生了数百万条短序列,这些序列映射到参考基因组后,可提供基因组、转录组和表观基因组水平的生物学见解。通常,正确映射到参考基因组的读取量在 70%到 90%之间,在某些情况下,仍会有一部分未映射的序列。这种“未对齐”可能归因于低质量碱基或样本读取与参考基因组之间的序列差异。研究未映射读取的来源对于更好地评估整个实验的质量并检查是否存在外源核酸的下游或上游“污染”至关重要。
在这里,我们提出了 DecontaMiner,这是一种用于揭示未映射读取中存在污染序列的工具。它使用减法方法来识别细菌、真菌和病毒基因组污染。DecontaMiner 生成多个输出文件来跟踪所有处理的读取,并提供其特征的完整报告。对微生物基因组的高质量匹配进行计数并在样本之间进行比较。DecontaMiner 构建了一个包含摘要统计信息和图表的离线 HTML 页面。后者使用最先进的 D3 JavaScript 库获得。DecontaMiner 主要用于检测人类 RNA-Seq 数据中的污染。该软件可免费在 http://www-labgtp.na.icar.cnr.it/decontaminer 获得。
DecontaMiner 是一种设计和开发用于调查未映射 NGS 数据中存在污染序列的工具。它可以提示测序样本中存在污染生物,这些生物可能来自实验室污染或其生物来源,在这两种情况下,都值得进一步调查和实验验证。DecontaMiner 的新颖之处主要在于它与 NGS 数据分析的标准程序的轻松集成,同时提供了一个完整、可靠和自动化的流程。