Institute for Physiology and Pathophysiology, Vegetative Physiology Group, University of Marburg, 35037 Marburg, Germany.
J Biol Chem. 2011 Apr 22;286(16):13977-84. doi: 10.1074/jbc.M111.227884. Epub 2011 Mar 1.
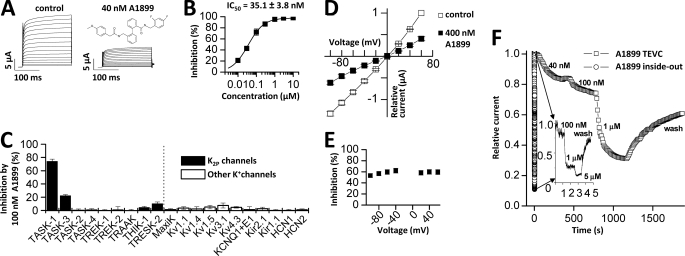
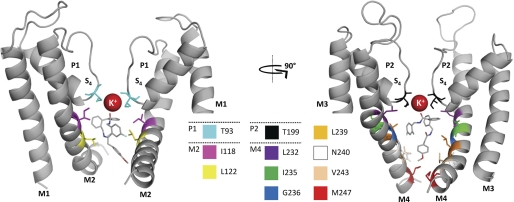
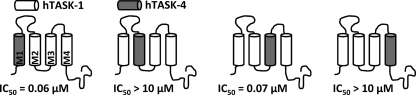
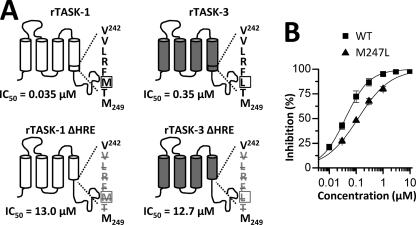
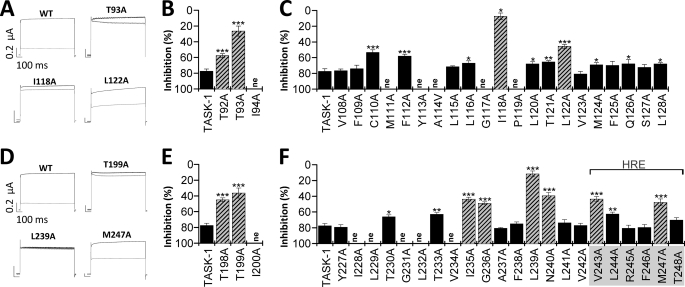
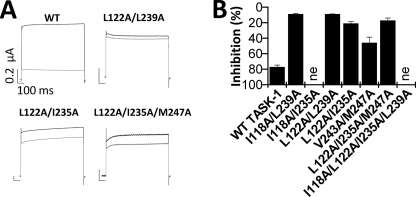
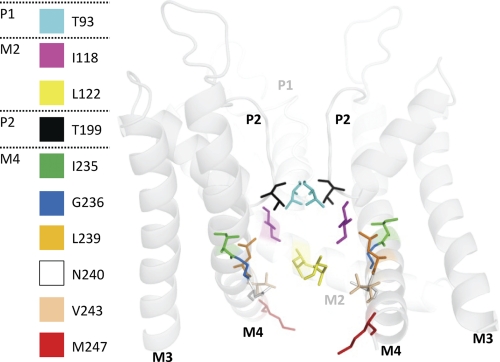
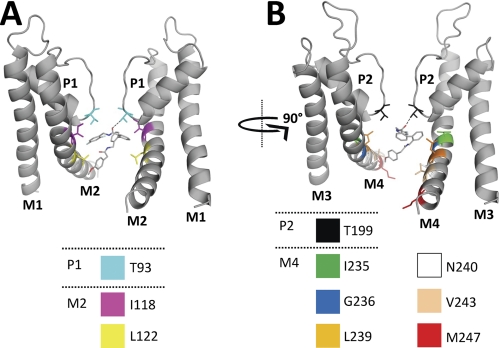
Two-pore domain potassium (K(2P)) channels play a key role in setting the membrane potential of excitable cells. Despite their role as putative targets for drugs and general anesthetics, little is known about the structure and the drug binding site of K(2P) channels. We describe A1899 as a potent and highly selective blocker of the K(2P) channel TASK-1. As A1899 acts as an open-channel blocker and binds to residues forming the wall of the central cavity, the drug was used to further our understanding of the channel pore. Using alanine mutagenesis screens, we have identified residues in both pore loops, the M2 and M4 segments, and the halothane response element to form the drug binding site of TASK-1. Our experimental data were used to validate a K(2P) open-pore homology model of TASK-1, providing structural insights for future rational design of drugs targeting K(2P) channels.
双孔钾(K(2P))通道在调节可兴奋细胞的膜电位方面起着关键作用。尽管它们被认为是药物和全身麻醉剂的潜在靶点,但对于 K(2P)通道的结构和药物结合部位知之甚少。我们将 A1899 描述为 TASK-1 K(2P)通道的有效且高度选择性的阻断剂。由于 A1899 作为开放通道阻断剂并结合形成中央腔壁的残基,因此该药物可用于进一步了解通道孔。通过丙氨酸诱变筛选,我们已经确定了位于两个孔环、M2 和 M4 片段以及卤烷反应元件中的残基,以形成 TASK-1 的药物结合部位。我们的实验数据用于验证 TASK-1 的 K(2P)开放孔同源模型,为未来针对 K(2P)通道的药物的合理设计提供结构见解。