Department of Computer Science, University College London, London, United Kingdom.
PLoS One. 2011 Feb 28;6(2):e16774. doi: 10.1371/journal.pone.0016774.
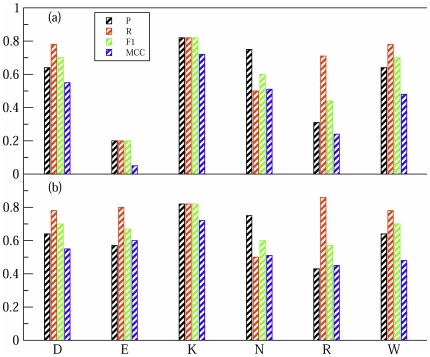
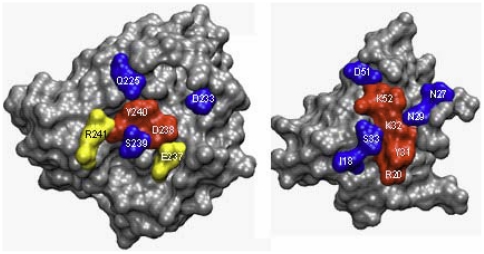
Protein-protein interactions are critically dependent on just a few 'hot spot' residues at the interface. Hot spots make a dominant contribution to the free energy of binding and they can disrupt the interaction if mutated to alanine. Here, we present HSPred, a support vector machine(SVM)-based method to predict hot spot residues, given the structure of a complex. HSPred represents an improvement over a previously described approach (Lise et al, BMC Bioinformatics 2009, 10:365). It achieves higher accuracy by treating separately predictions involving either an arginine or a glutamic acid residue. These are the amino acid types on which the original model did not perform well. We have therefore developed two additional SVM classifiers, specifically optimised for these cases. HSPred reaches an overall precision and recall respectively of 61% and 69%, which roughly corresponds to a 10% improvement. An implementation of the described method is available as a web server at http://bioinf.cs.ucl.ac.uk/hspred. It is free to non-commercial users.
蛋白质-蛋白质相互作用严重依赖于界面上的几个“热点”残基。热点对结合自由能有很大的贡献,如果突变为丙氨酸,它们可能会破坏相互作用。在这里,我们提出了 HSPred,这是一种基于支持向量机(SVM)的方法,用于预测热点残基,给定复合物的结构。HSPred 比之前描述的方法(Lise 等人,BMC Bioinformatics 2009,10:365)有所改进。它通过分别处理涉及精氨酸或谷氨酸残基的预测来实现更高的准确性。这些是原始模型表现不佳的氨基酸类型。因此,我们开发了两个额外的 SVM 分类器,专门针对这些情况进行了优化。HSPred 的总体精度和召回率分别为 61%和 69%,大致相当于提高了 10%。描述的方法的实现可以作为一个网络服务器在 http://bioinf.cs.ucl.ac.uk/hspred 上获得。它对非商业用户是免费的。