Département de Sciences biologiques, Université de Montréal, C.P. 6128, Succ, Centre-ville, Montréal, Québec H3C3J7, Canada.
BMC Evol Biol. 2011 Mar 9;11:64. doi: 10.1186/1471-2148-11-64.
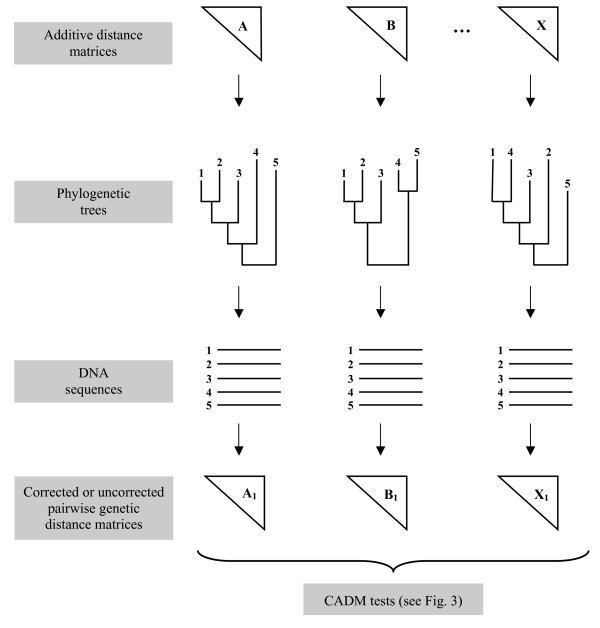
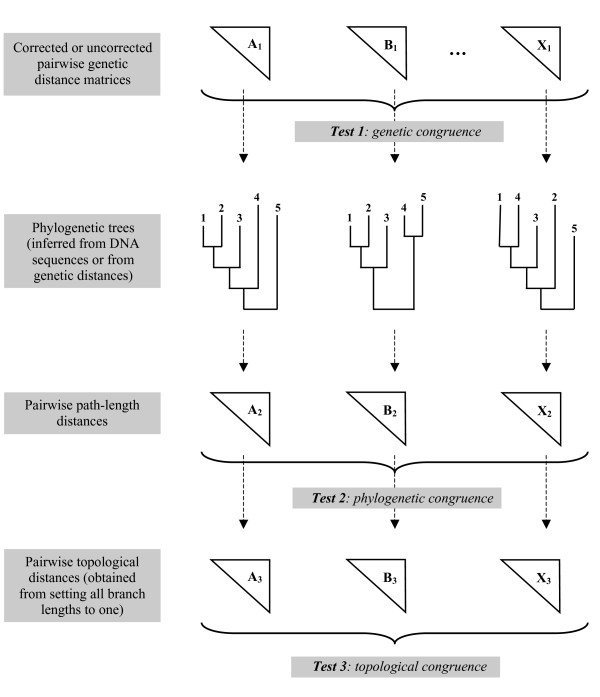
CADM is a statistical test used to estimate the level of Congruence Among Distance Matrices. It has been shown in previous studies to have a correct rate of type I error and good power when applied to dissimilarity matrices and to ultrametric distance matrices. Contrary to most other tests of incongruence used in phylogenetic analysis, the null hypothesis of the CADM test assumes complete incongruence of the phylogenetic trees instead of congruence. In this study, we performed computer simulations to assess the type I error rate and power of the test. It was applied to additive distance matrices representing phylogenies and to genetic distance matrices obtained from nucleotide sequences of different lengths that were simulated on randomly generated trees of varying sizes, and under different evolutionary conditions.
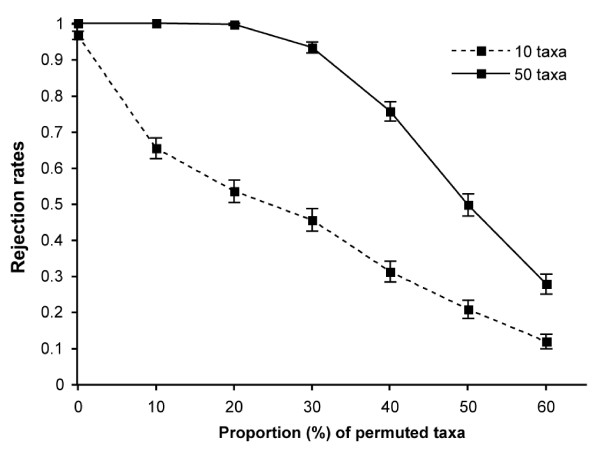
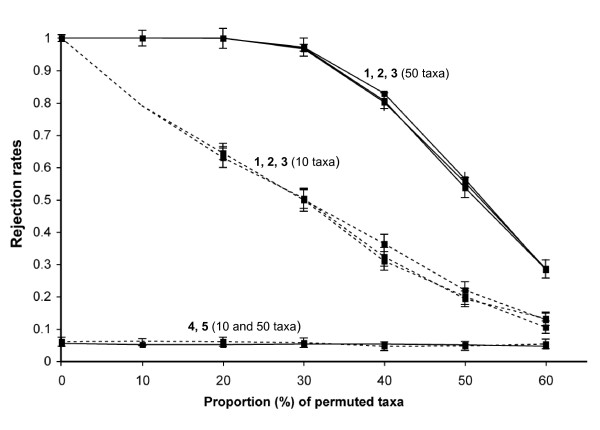
Our results showed that the test has an accurate type I error rate and good power. As expected, power increased with the number of objects (i.e., taxa), the number of partially or completely congruent matrices and the level of congruence among distance matrices.
Based on our results, we suggest that CADM is an excellent candidate to test for congruence and, when present, to estimate its level in phylogenomic studies where numerous genes are analysed simultaneously.
CADM 是一种统计检验方法,用于估计距离矩阵之间的一致性程度。先前的研究表明,该检验方法在应用于不相似性矩阵和超度量距离矩阵时,具有正确的Ⅰ类错误率和良好的功效。与系统发育分析中使用的大多数其他不一致性检验方法不同,CADM 检验的零假设假设系统发育树完全不一致,而不是一致。在这项研究中,我们进行了计算机模拟,以评估检验的Ⅰ类错误率和功效。它应用于代表系统发育的加性距离矩阵,以及从不同长度的核苷酸序列获得的遗传距离矩阵,这些矩阵是在大小不同、进化条件不同的随机生成的树上模拟的。
我们的结果表明,该检验具有准确的Ⅰ类错误率和良好的功效。正如预期的那样,功效随着对象(即分类单元)的数量、部分或完全一致的矩阵的数量以及距离矩阵之间一致性的水平而增加。
基于我们的结果,我们建议 CADM 是一个极好的候选者,可以用于检验一致性,并且在同时分析多个基因的基因组研究中,当存在一致性时,可以估计其水平。