DoD Biotechnology High Performance Computing Software Applications Institute, Telemedicine and Advanced Technology Research Center, U.S. Army Medical Research and Materiel Command, Ft. Detrick, Maryland, United States of America.
PLoS One. 2011 Mar 7;6(3):e17469. doi: 10.1371/journal.pone.0017469.
The annotation of genomes from next-generation sequencing platforms needs to be rapid, high-throughput, and fully integrated and automated. Although a few Web-based annotation services have recently become available, they may not be the best solution for researchers that need to annotate a large number of genomes, possibly including proprietary data, and store them locally for further analysis. To address this need, we developed a standalone software application, the Annotation of microbial Genome Sequences (AGeS) system, which incorporates publicly available and in-house-developed bioinformatics tools and databases, many of which are parallelized for high-throughput performance.
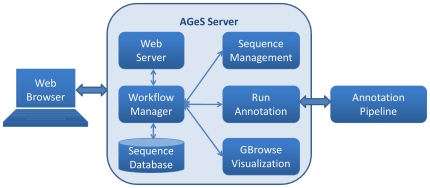
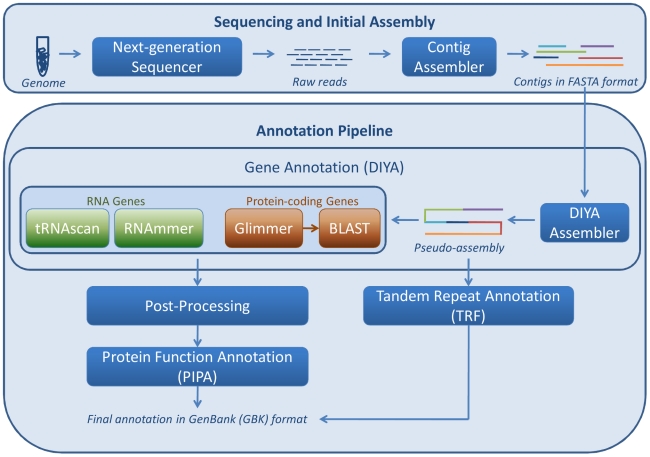
The AGeS system supports three main capabilities. The first is the storage of input contig sequences and the resulting annotation data in a central, customized database. The second is the annotation of microbial genomes using an integrated software pipeline, which first analyzes contigs from high-throughput sequencing by locating genomic regions that code for proteins, RNA, and other genomic elements through the Do-It-Yourself Annotation (DIYA) framework. The identified protein-coding regions are then functionally annotated using the in-house-developed Pipeline for Protein Annotation (PIPA). The third capability is the visualization of annotated sequences using GBrowse. To date, we have implemented these capabilities for bacterial genomes. AGeS was evaluated by comparing its genome annotations with those provided by three other methods. Our results indicate that the software tools integrated into AGeS provide annotations that are in general agreement with those provided by the compared methods. This is demonstrated by a >94% overlap in the number of identified genes, a significant number of identical annotated features, and a >90% agreement in enzyme function predictions.
下一代测序平台的基因组注释需要快速、高通量、完全集成和自动化。尽管最近出现了一些基于网络的注释服务,但对于需要注释大量基因组(可能包括专有数据)并将其存储在本地进行进一步分析的研究人员来说,它们可能不是最佳解决方案。为了满足这一需求,我们开发了一个独立的软件应用程序,即微生物基因组序列注释(AGeS)系统,该系统集成了公开和内部开发的生物信息学工具和数据库,其中许多工具和数据库都经过并行化处理,以实现高通量性能。
AGeS 系统支持三个主要功能。第一个功能是在中央自定义数据库中存储输入的连续序列和生成的注释数据。第二个功能是使用集成的软件管道注释微生物基因组,该管道首先通过 DIYA 框架定位编码蛋白质、RNA 和其他基因组元件的基因组区域,对高通量测序的连续序列进行分析。然后,使用内部开发的蛋白质注释管道(PIPA)对识别的蛋白质编码区域进行功能注释。第三个功能是使用 GBrowse 可视化注释序列。迄今为止,我们已经为细菌基因组实现了这些功能。通过将 AGeS 的基因组注释与其他三种方法的注释进行比较,对其进行了评估。我们的结果表明,集成到 AGeS 中的软件工具提供的注释通常与比较方法提供的注释一致。这一点通过以下几个方面得到证明:鉴定基因的数量有超过 94%的重叠,相同注释特征的数量非常多,酶功能预测的一致性超过 90%。