Pagani Lucia, Eckert Anne
Neurobiology Laboratory for Brain Aging and Mental Health, Psychiatric University Clinics, University of Basel, Wilhelm Klein-Straße 27, 4012 Basel, Switzerland.
Int J Alzheimers Dis. 2011 Mar 15;2011:925050. doi: 10.4061/2011/925050.
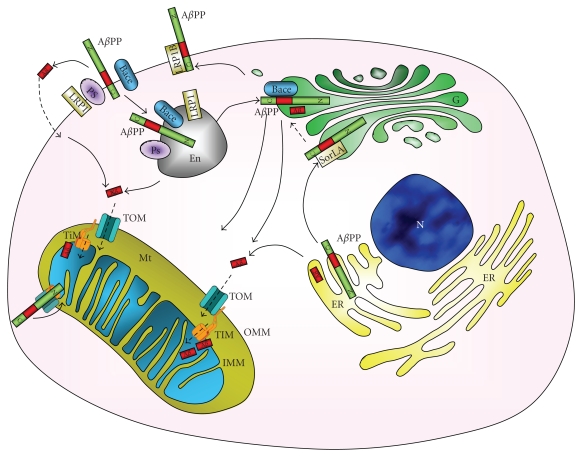
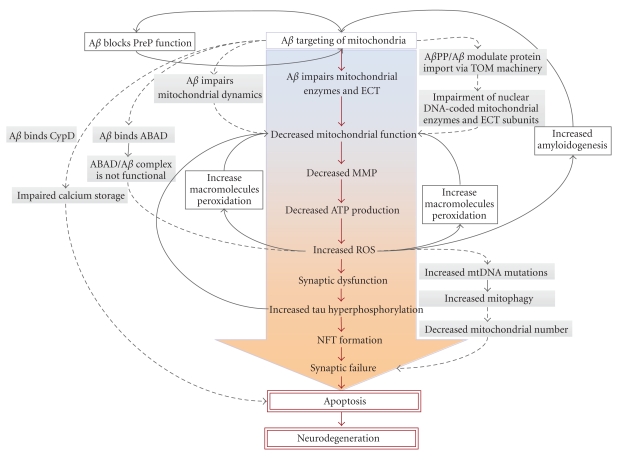
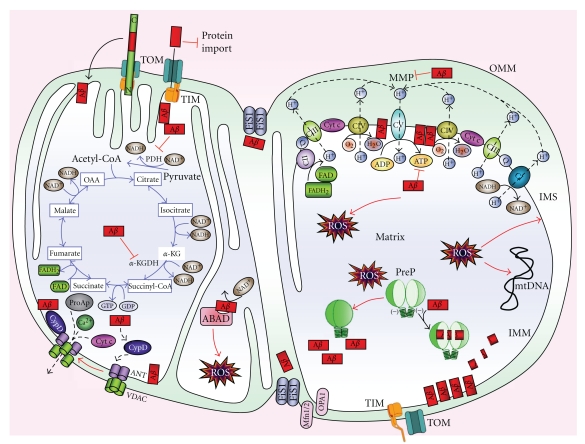
Mitochondrial dysfunction is a hallmark of amyloid-beta(Aβ)-induced neuronal toxicity in Alzheimer's disease (AD). The recent emphasis on the intracellular biology of Aβ and its precursor protein (AβPP) has led researchers to consider the possibility that mitochondria-associated and/or intramitochondrial Aβ may directly cause neurotoxicity. In this paper, we will outline current knowledge of the intracellular localization of both Aβ and AβPP addressing the question of how Aβ can access mitochondria. Moreover, we summarize evidence from AD postmortem brain as well as cellular and animal AD models showing that Aβ triggers mitochondrial dysfunction through a number of pathways such as impairment of oxidative phosphorylation, elevation of reactive oxygen species (ROS) production, alteration of mitochondrial dynamics, and interaction with mitochondrial proteins. In particular, we focus on Aβ interaction with different mitochondrial targets including the outer mitochondrial membrane, intermembrane space, inner mitochondrial membrane, and the matrix. Thus, this paper establishes a modified model of the Alzheimer cascade mitochondrial hypothesis.
线粒体功能障碍是阿尔茨海默病(AD)中淀粉样β蛋白(Aβ)诱导的神经元毒性的一个标志。最近对Aβ及其前体蛋白(AβPP)细胞内生物学的重视,促使研究人员考虑线粒体相关和/或线粒体内Aβ可能直接导致神经毒性的可能性。在本文中,我们将概述目前关于Aβ和AβPP细胞内定位的知识,探讨Aβ如何进入线粒体的问题。此外,我们总结了来自AD死后大脑以及细胞和动物AD模型的证据,表明Aβ通过多种途径触发线粒体功能障碍,如氧化磷酸化受损、活性氧(ROS)生成增加、线粒体动力学改变以及与线粒体蛋白相互作用。特别是,我们关注Aβ与不同线粒体靶点的相互作用,包括线粒体外膜、膜间隙、线粒体内膜和线粒体基质。因此,本文建立了阿尔茨海默病级联线粒体假说的修正模型。