Animal Breeding and Genomics Centre, Wageningen University, Wageningen, The Netherlands.
PLoS One. 2011 Apr 4;6(4):e14782. doi: 10.1371/journal.pone.0014782.
Artificial selection has caused rapid evolution in domesticated species. The identification of selection footprints across domesticated genomes can contribute to uncover the genetic basis of phenotypic diversity.
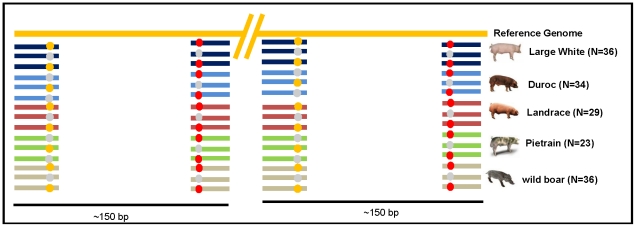
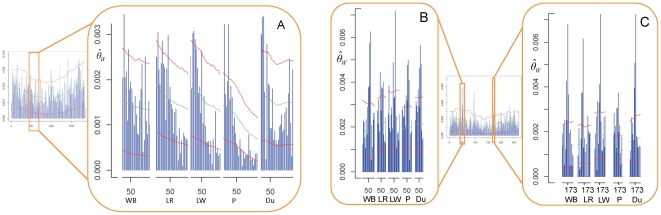
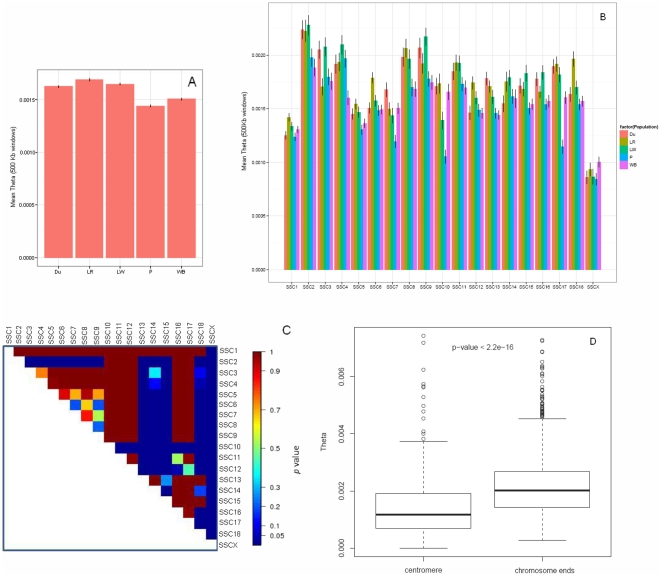
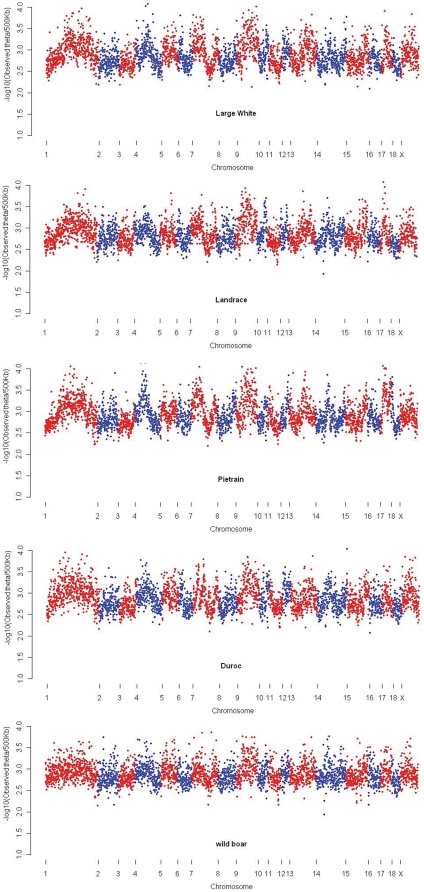
METHODOLOGY/MAIN FINDINGS: Genome wide footprints of pig domestication and selection were identified using massive parallel sequencing of pooled reduced representation libraries (RRL) representing ∼2% of the genome from wild boar and four domestic pig breeds (Large White, Landrace, Duroc and Pietrain) which have been under strong selection for muscle development, growth, behavior and coat color. Using specifically developed statistical methods that account for DNA pooling, low mean sequencing depth, and sequencing errors, we provide genome-wide estimates of nucleotide diversity and genetic differentiation in pig. Widespread signals suggestive of positive and balancing selection were found and the strongest signals were observed in Pietrain, one of the breeds most intensively selected for muscle development. Most signals were population-specific but affected genomic regions which harbored genes for common biological categories including coat color, brain development, muscle development, growth, metabolism, olfaction and immunity. Genetic differentiation in regions harboring genes related to muscle development and growth was higher between breeds than between a given breed and the wild boar.
CONCLUSIONS/SIGNIFICANCE: These results, suggest that although domesticated breeds have experienced similar selective pressures, selection has acted upon different genes. This might reflect the multiple domestication events of European breeds or could be the result of subsequent introgression of Asian alleles. Overall, it was estimated that approximately 7% of the porcine genome has been affected by selection events. This study illustrates that the massive parallel sequencing of genomic pools is a cost-effective approach to identify footprints of selection.
人工选择导致了家养物种的快速进化。在驯化基因组中识别选择足迹可以帮助揭示表型多样性的遗传基础。
方法/主要发现:利用大量平行测序的 pooled reduced representation libraries (RRL),对来自野猪和四个家猪品种(长白猪、兰德瑞斯猪、杜洛克猪和皮特兰猪)的基因组进行了驯化和选择的全基因组足迹识别。这些品种在肌肉发育、生长、行为和毛色方面受到了强烈的选择。我们使用专门开发的统计方法,考虑了 DNA 池化、平均测序深度低和测序错误,提供了猪的核苷酸多样性和遗传分化的全基因组估计。发现了广泛的正向选择和平衡选择的信号,其中最强的信号出现在皮特兰猪中,这是肌肉发育选择最强烈的品种之一。大多数信号是种群特异性的,但影响了包含毛色、脑发育、肌肉发育、生长、代谢、嗅觉和免疫等常见生物学类别的基因的基因组区域。与肌肉发育和生长相关的基因所在区域的遗传分化在品种之间高于给定品种和野猪之间。
结论/意义:这些结果表明,尽管家养品种经历了相似的选择压力,但选择作用于不同的基因。这可能反映了欧洲品种的多次驯化事件,也可能是亚洲等位基因随后渗入的结果。总体而言,估计约有 7%的猪基因组受到了选择事件的影响。这项研究表明,大规模平行测序基因组池是一种经济有效的方法,可以识别选择的足迹。