Wang Zhen, Chen Qiang, Yang Yumei, Yang Hongjie, He Pengfei, Zhang Zhe, Chen Zhenliang, Liao Rongrong, Tu Yingying, Zhang Xiangzhe, Wang Qishan, Pan Yuchun
Department of Animal Science, School of Agriculture and Biology, Shanghai Jiao Tong University, Shanghai, 200240, China; Shanghai Key Laboratory of Veterinary Biotechnology, Shanghai, 200240, China.
Anim Genet. 2014 Dec;45(6):808-16. doi: 10.1111/age.12229. Epub 2014 Oct 19.
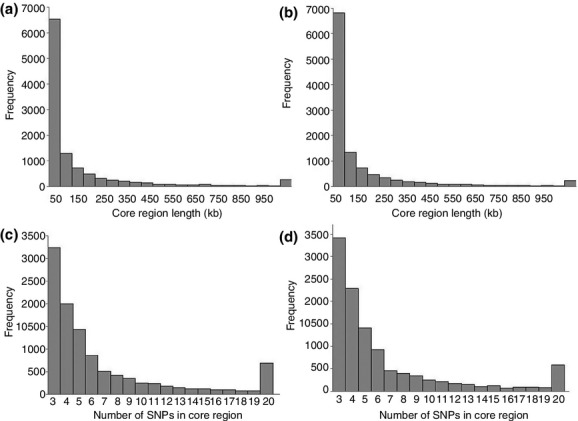
Pigs have experienced dramatic selection due to domestication, which has led to many different phenotypes when compared to their wild counterparts, especially in the last several decades. Currently, genome-wide scans in both cattle and humans showing positive selection footprints have been investigated. However, few studies have focused on porcine selection footprints, particularly on a genome-wide scale. Surveying for selection footprints across porcine genomes can be quite valuable for revealing the genetic mechanisms of phenotypic diversity. Here, we employed a medium sequencing depth (5-20x/site per individual, on average) approach called genotyping by genome reducing and sequencing (GGRS) to detect genome-wide selection signatures of two domestic pig breeds (Yorkshire and Landrace) that have been under intensive selection for traits of muscle development, growth and behavior. The relative extended haplotype homozygosity test, which identifies selection signatures by measuring the characteristics of haplotypes' frequency distribution within a single population, was also applied to identify potential positively selected regions. As a result, signatures of positive selection were found in each breed. However, most selection signatures were population specific and related to genomic regions containing genes for biological categories including brain development, metabolism, growth and olfaction. Furthermore, the result of the gene set enrichment analysis indicated that selected regions of the two breeds presented a different over-representation of genes in the Gene Ontology annotations and Kyoto Encyclopedia of Genes and Genomes pathways. Our results revealed a genome-wide map of selection footprints in pigs and may help us better understand the mechanisms of selection in pig breeding.
由于驯化,猪经历了剧烈的选择,与它们的野生同类相比,这导致了许多不同的表型,尤其是在过去几十年。目前,已经对牛和人类中显示正选择足迹的全基因组扫描进行了研究。然而,很少有研究关注猪的选择足迹,特别是在全基因组规模上。在猪基因组中寻找选择足迹对于揭示表型多样性的遗传机制可能非常有价值。在这里,我们采用了一种中等测序深度(平均每个个体每个位点5-20倍)的方法,即基因组缩减测序基因分型(GGRS),来检测两个经过高强度选择以获得肌肉发育、生长和行为性状的家猪品种(约克夏和长白)的全基因组选择特征。相对扩展单倍型纯合性测试,通过测量单一种群内单倍型频率分布的特征来识别选择特征,也被用于识别潜在的正选择区域。结果,在每个品种中都发现了正选择特征。然而,大多数选择特征是种群特异性的,并且与包含涉及大脑发育、代谢、生长和嗅觉等生物学类别的基因的基因组区域相关。此外,基因集富集分析的结果表明,两个品种的选择区域在基因本体注释和京都基因与基因组百科全书途径中呈现出不同的基因过度代表性。我们的结果揭示了猪全基因组选择足迹图谱,可能有助于我们更好地理解猪育种中的选择机制。