Department of Physics and Astronomy, Rutgers University, Piscataway, New Jersey, USA.
BMC Bioinformatics. 2010 Jun 24;11:345. doi: 10.1186/1471-2105-11-345.
High throughput sequencing (HTS) platforms produce gigabases of short read (<100 bp) data per run. While these short reads are adequate for resequencing applications, de novo assembly of moderate size genomes from such reads remains a significant challenge. These limitations could be partially overcome by utilizing mate pair technology, which provides pairs of short reads separated by a known distance along the genome.
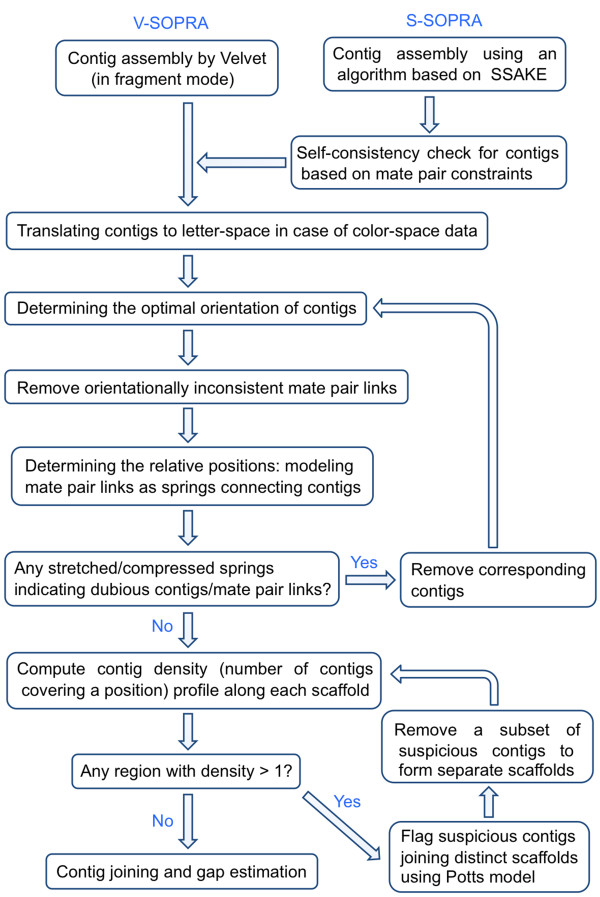
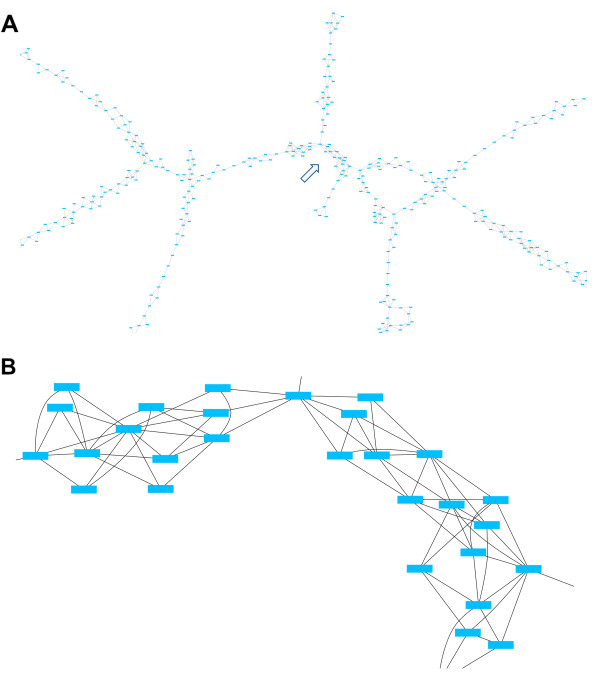
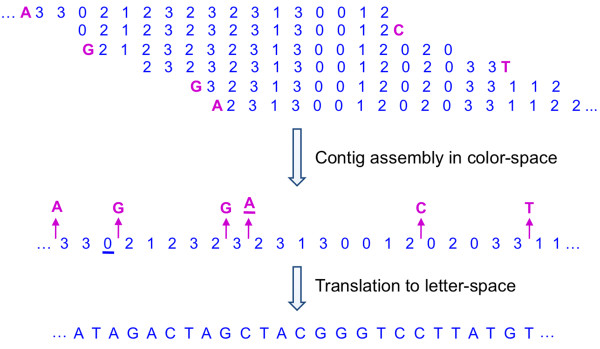
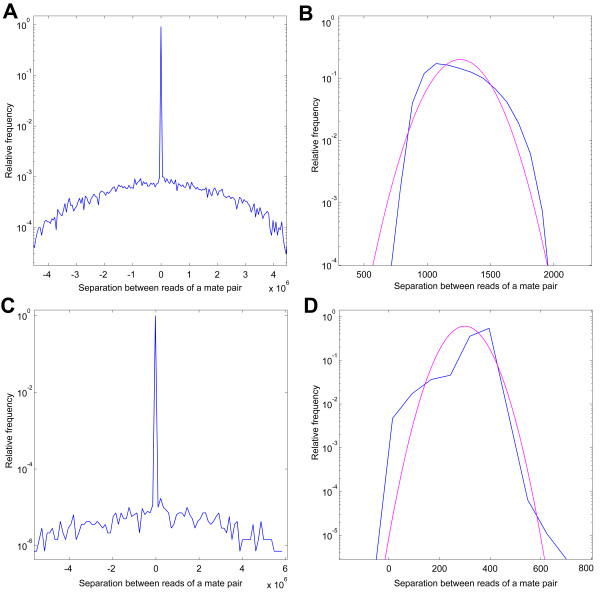
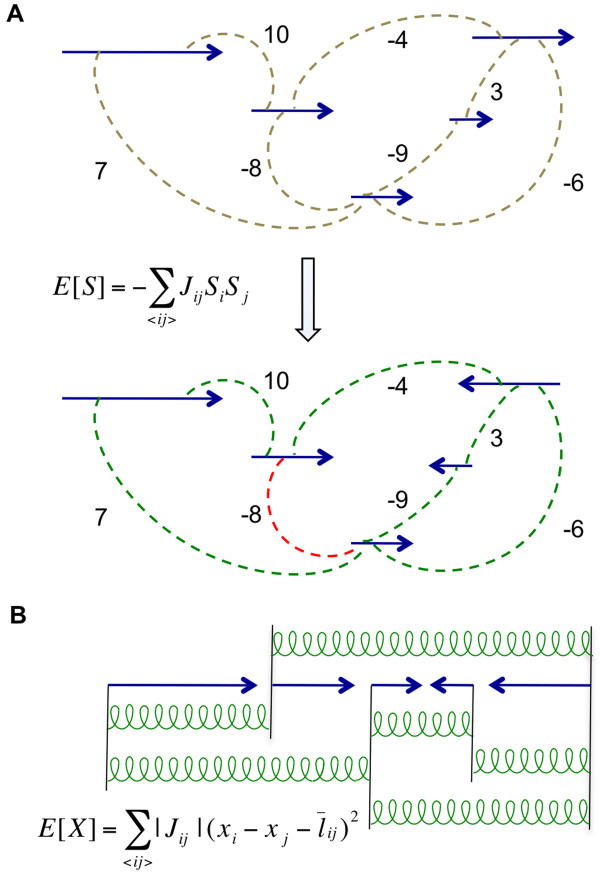
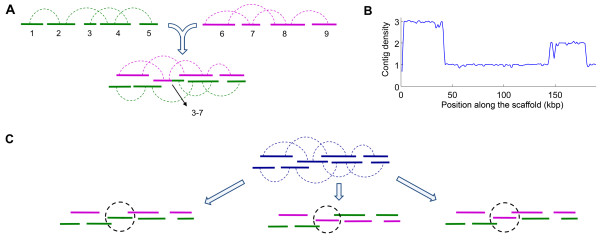
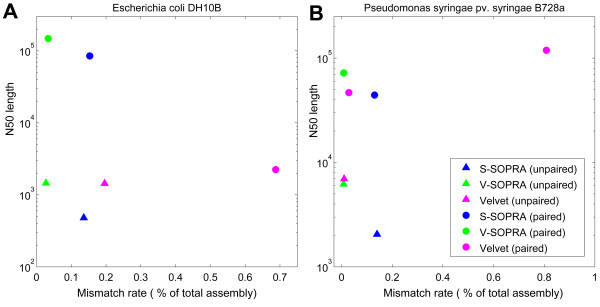
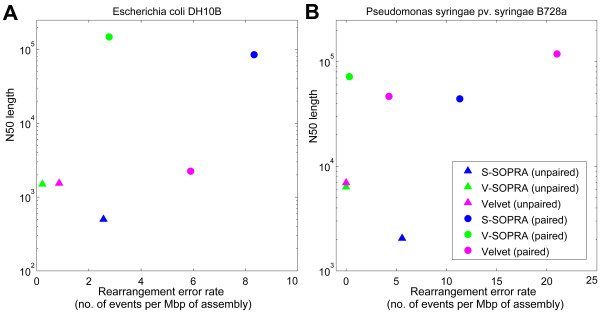
We have developed SOPRA, a tool designed to exploit the mate pair/paired-end information for assembly of short reads. The main focus of the algorithm is selecting a sufficiently large subset of simultaneously satisfiable mate pair constraints to achieve a balance between the size and the quality of the output scaffolds. Scaffold assembly is presented as an optimization problem for variables associated with vertices and with edges of the contig connectivity graph. Vertices of this graph are individual contigs with edges drawn between contigs connected by mate pairs. Similar graph problems have been invoked in the context of shotgun sequencing and scaffold building for previous generation of sequencing projects. However, given the error-prone nature of HTS data and the fundamental limitations from the shortness of the reads, the ad hoc greedy algorithms used in the earlier studies are likely to lead to poor quality results in the current context. SOPRA circumvents this problem by treating all the constraints on equal footing for solving the optimization problem, the solution itself indicating the problematic constraints (chimeric/repetitive contigs, etc.) to be removed. The process of solving and removing of constraints is iterated till one reaches a core set of consistent constraints. For SOLiD sequencer data, SOPRA uses a dynamic programming approach to robustly translate the color-space assembly to base-space. For assessing the quality of an assembly, we report the no-match/mismatch error rate as well as the rates of various rearrangement errors.
Applying SOPRA to real data from bacterial genomes, we were able to assemble contigs into scaffolds of significant length (N50 up to 200 Kb) with very few errors introduced in the process. In general, the methodology presented here will allow better scaffold assemblies of any type of mate pair sequencing data.
高通量测序(HTS)平台在每次运行时都会产生千兆字节的短读(<100bp)数据。虽然这些短读足以满足重测序应用,但从这些读段从头组装中等大小的基因组仍然是一个重大挑战。通过利用配对末端技术,这些限制可以部分克服,该技术提供了基因组上已知距离的一对短读段。
我们开发了 SOPRA,这是一种设计用于利用配对末端/成对末端信息进行短读段组装的工具。该算法的主要重点是选择一个足够大的同时满足的配对末端约束子集,以在输出支架的大小和质量之间取得平衡。支架组装被呈现为与顶点和连接图的边相关的变量的优化问题。该图的顶点是个体支架,边缘是通过配对末端连接的支架之间绘制的。在以前的测序项目中,已经在霰弹枪测序和支架构建的背景下调用了类似的图问题。然而,鉴于 HTS 数据的易错性质以及由于读段较短而带来的根本限制,在当前背景下,早期研究中使用的特定贪婪算法可能会导致质量较差的结果。SOPRA 通过平等对待所有约束来解决优化问题,从而避免了这个问题,解决方案本身表明需要去除有问题的约束(嵌合/重复支架等)。该约束的解决和去除过程一直迭代,直到达到一个核心的一致约束集。对于 SOLiD 测序仪数据,SOPRA 使用动态规划方法来稳健地将颜色空间组装转换为碱基空间。为了评估组装的质量,我们报告无匹配/不匹配错误率以及各种重排错误率。
将 SOPRA 应用于来自细菌基因组的真实数据,我们能够将支架组装成具有显著长度的支架(N50 高达 200kb),并且在组装过程中引入的错误很少。一般来说,这里提出的方法学将允许更好地组装任何类型的配对末端测序数据的支架。