Dept, of Computer Science and Engineering, UC San Diego, La Jolla, CA, USA.
BMC Genomics. 2010 Jun 18;11:385. doi: 10.1186/1471-2164-11-385.
Massively parallel DNA sequencing technologies have enabled the sequencing of several individual human genomes. These technologies are also being used in novel ways for mRNA expression profiling, genome-wide discovery of transcription-factor binding sites, small RNA discovery, etc. The multitude of sequencing platforms, each with their unique characteristics, pose a number of design challenges, regarding the technology to be used and the depth of sequencing required for a particular sequencing application. Here we describe a number of analytical and empirical results to address design questions for two applications: detection of structural variations from paired-end sequencing and estimating mRNA transcript abundance.
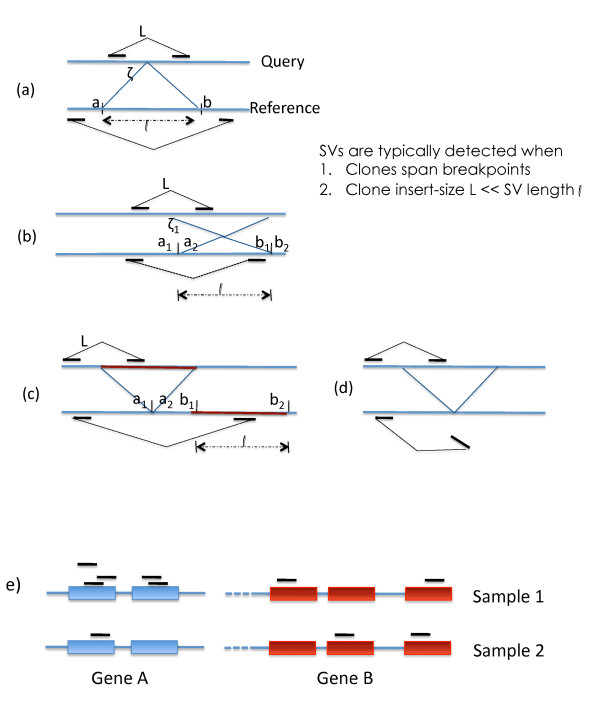
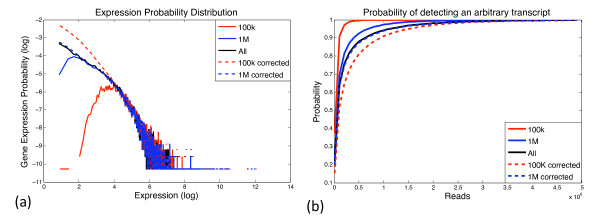
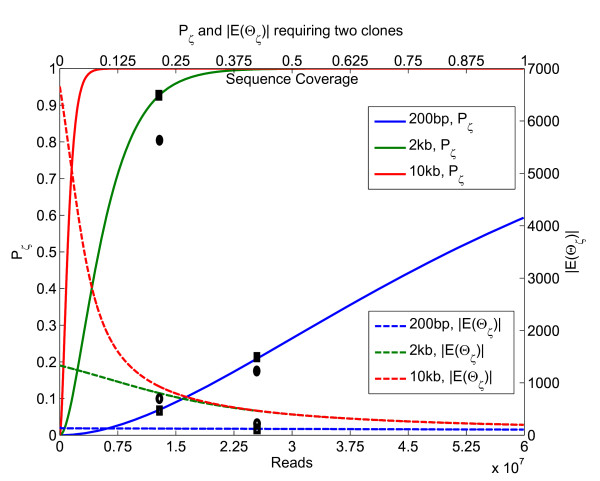
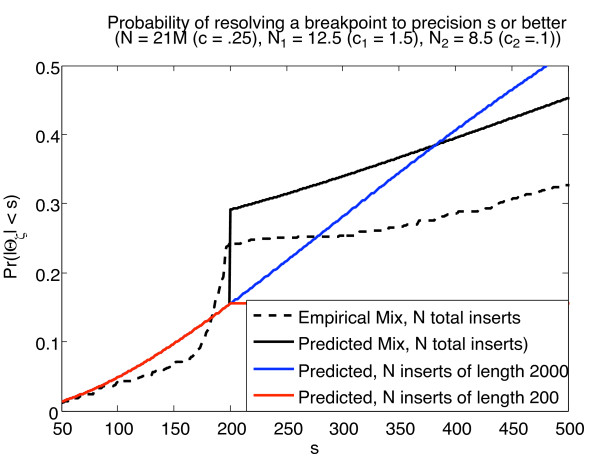
For structural variation, our results provide explicit trade-offs between the detection and resolution of rearrangement breakpoints, and the optimal mix of paired-read insert lengths. Specifically, we prove that optimal detection and resolution of breakpoints is achieved using a mix of exactly two insert library lengths. Furthermore, we derive explicit formulae to determine these insert length combinations, enabling a 15% improvement in breakpoint detection at the same experimental cost. On empirical short read data, these predictions show good concordance with Illumina 200 bp and 2 Kbp insert length libraries. For transcriptome sequencing, we determine the sequencing depth needed to detect rare transcripts from a small pilot study. With only 1 Million reads, we derive corrections that enable almost perfect prediction of the underlying expression probability distribution, and use this to predict the sequencing depth required to detect low expressed genes with greater than 95% probability.
Together, our results form a generic framework for many design considerations related to high-throughput sequencing. We provide software tools http://bix.ucsd.edu/projects/NGS-DesignTools to derive platform independent guidelines for designing sequencing experiments (amount of sequencing, choice of insert length, mix of libraries) for novel applications of next generation sequencing.
大规模平行 DNA 测序技术已经使得对几个个体人类基因组进行测序成为可能。这些技术也正在以新颖的方式用于 mRNA 表达谱分析、全基因组转录因子结合位点发现、小 RNA 发现等。多种测序平台,每个平台都具有独特的特点,在要使用的技术和特定测序应用所需的测序深度方面都带来了一些设计挑战。在这里,我们描述了一些分析和经验结果,以解决两个应用程序的设计问题:从配对末端测序中检测结构变异和估计 mRNA 转录本丰度。
对于结构变异,我们的结果提供了在重排断点的检测和分辨率以及最佳配对读取插入长度组合之间的明确权衡。具体来说,我们证明使用恰好两种插入文库长度的混合可以实现断点的最佳检测和分辨率。此外,我们推导出了确定这些插入长度组合的显式公式,使在相同实验成本下,断点检测提高了 15%。在经验性短读数据上,这些预测与 Illumina 200bp 和 2Kbp 插入长度文库具有很好的一致性。对于转录组测序,我们从一个小的试点研究中确定了检测稀有转录本所需的测序深度。仅使用 100 万个读数,我们得出了校正值,这些校正值几乎可以完美地预测潜在的表达概率分布,并使用这些值来预测以 95%以上的概率检测低表达基因所需的测序深度。
总的来说,我们的结果为与高通量测序相关的许多设计考虑因素形成了通用框架。我们提供了软件工具 http://bix.ucsd.edu/projects/NGS-DesignTools,为下一代测序的新型应用程序设计测序实验(测序量、插入长度选择、文库混合)提供了平台独立的指导原则。