Functional Genome Analysis, Deutsches Krebsforschungszentrum, Im Neuenheimer Feld 580, 69120 Heidelberg, Germany.
Nucleic Acids Res. 2011 Jun;39(11):e77. doi: 10.1093/nar/gkr213. Epub 2011 Apr 12.
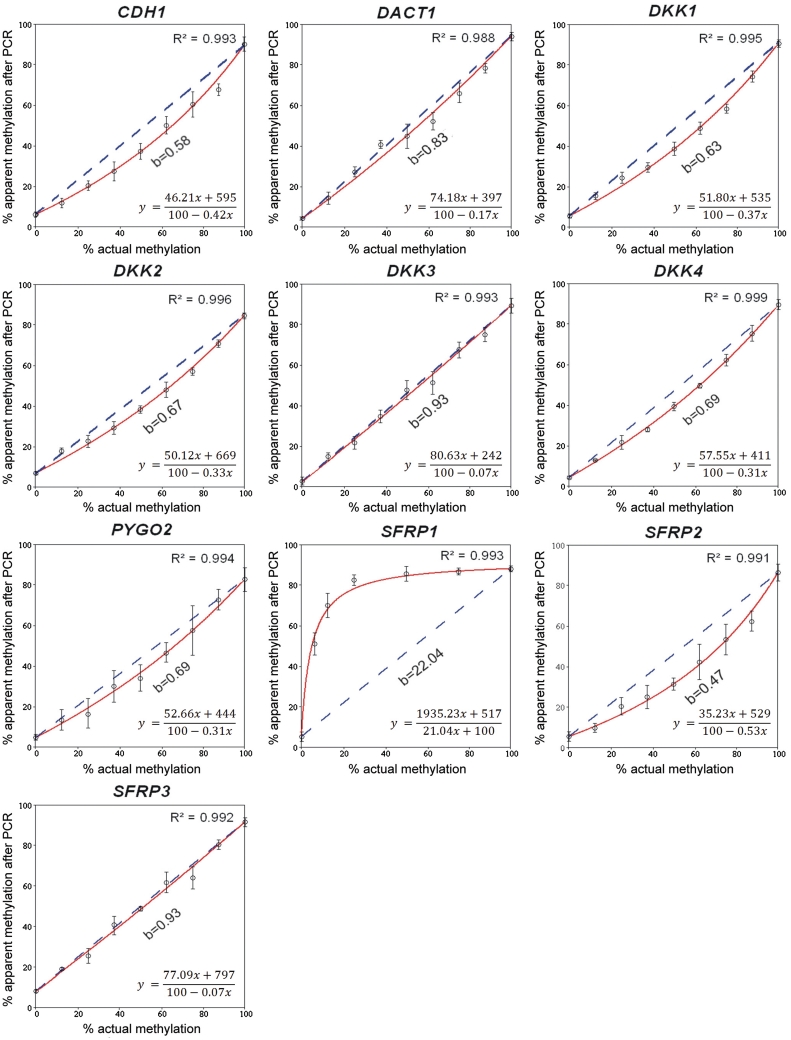
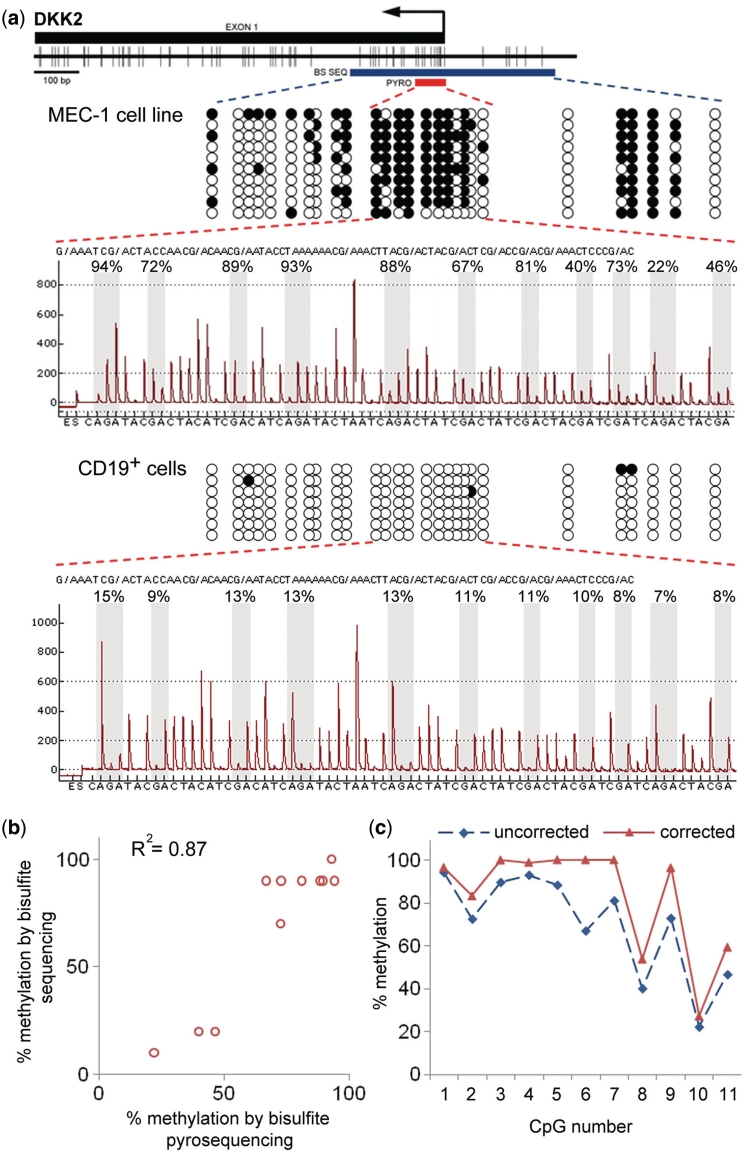
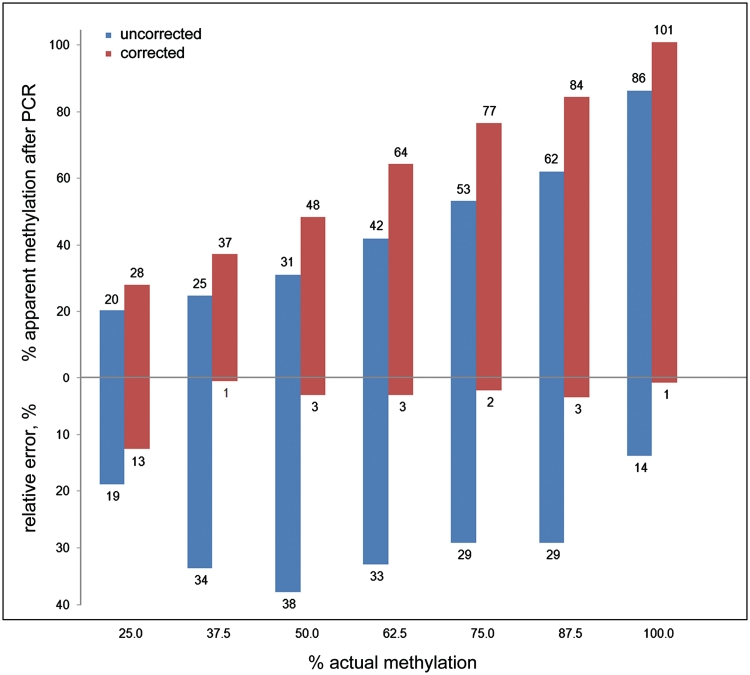
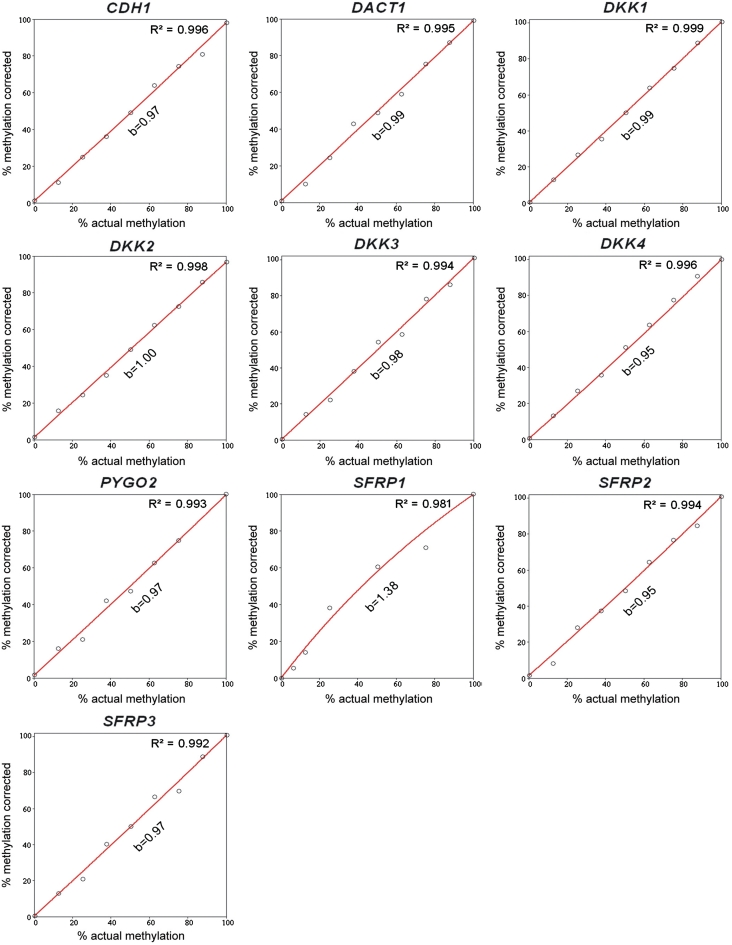
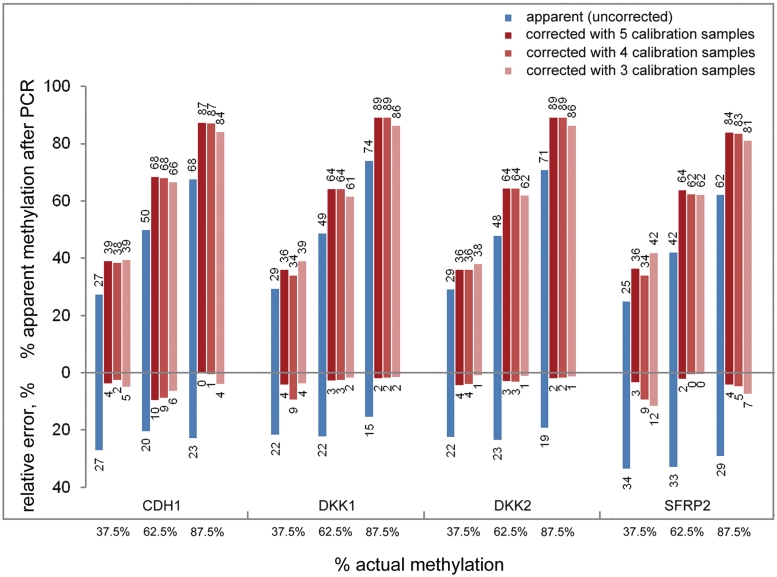
DNA methylation profiling has become an important aspect of biomedical molecular analysis. Polymerase chain reaction (PCR) amplification of bisulphite-treated DNA is a processing step that is common to many currently used methods of quantitative methylation analysis. Preferential amplification of unmethylated alleles-known as PCR-bias-may significantly affect the accuracy of quantification. To date, no universal experimental approach has been reported to overcome the problem. This study presents an effective method of correcting biased methylation data. The procedure includes a calibration performed in parallel to the analysis of the samples under investigation. DNA samples with defined degrees of methylation are analysed. The observed deviation of the experimental results from the expected values is used for calculating a regression curve. The equation of the best-fitting curve is then used for correction of the data obtained from the samples of interest. The process can be applied irrespective of the locus interrogated and the number of sites analysed, avoiding an optimization of the amplification conditions for each individual locus.
DNA 甲基化分析已成为生物医学分子分析的一个重要方面。亚硫酸氢盐处理的 DNA 的聚合酶链反应(PCR)扩增是许多目前使用的定量甲基化分析方法中常见的处理步骤。未甲基化等位基因的优先扩增——称为 PCR 偏倚——可能会显著影响定量的准确性。迄今为止,尚未报道任何通用的实验方法来克服该问题。本研究提出了一种纠正偏置甲基化数据的有效方法。该程序包括在分析研究样本的同时进行校准。分析具有定义程度甲基化的 DNA 样本。从观察到的实验结果与预期值的偏差中,计算出回归曲线。然后,使用最佳拟合曲线的方程来校正从感兴趣的样本中获得的数据。该过程可以应用于无论检测的基因座和分析的位点数量如何,避免了为每个单独的基因座优化扩增条件。