College of Information Science and Technology, Pennsylvania State University, State College, Pennsylvania, United States of America.
Institute for Computational and Data Science, Pennsylvania State University, State College, Pennsylvania, United States of America.
PLoS Comput Biol. 2021 Jul 6;17(7):e1009113. doi: 10.1371/journal.pcbi.1009113. eCollection 2021 Jul.
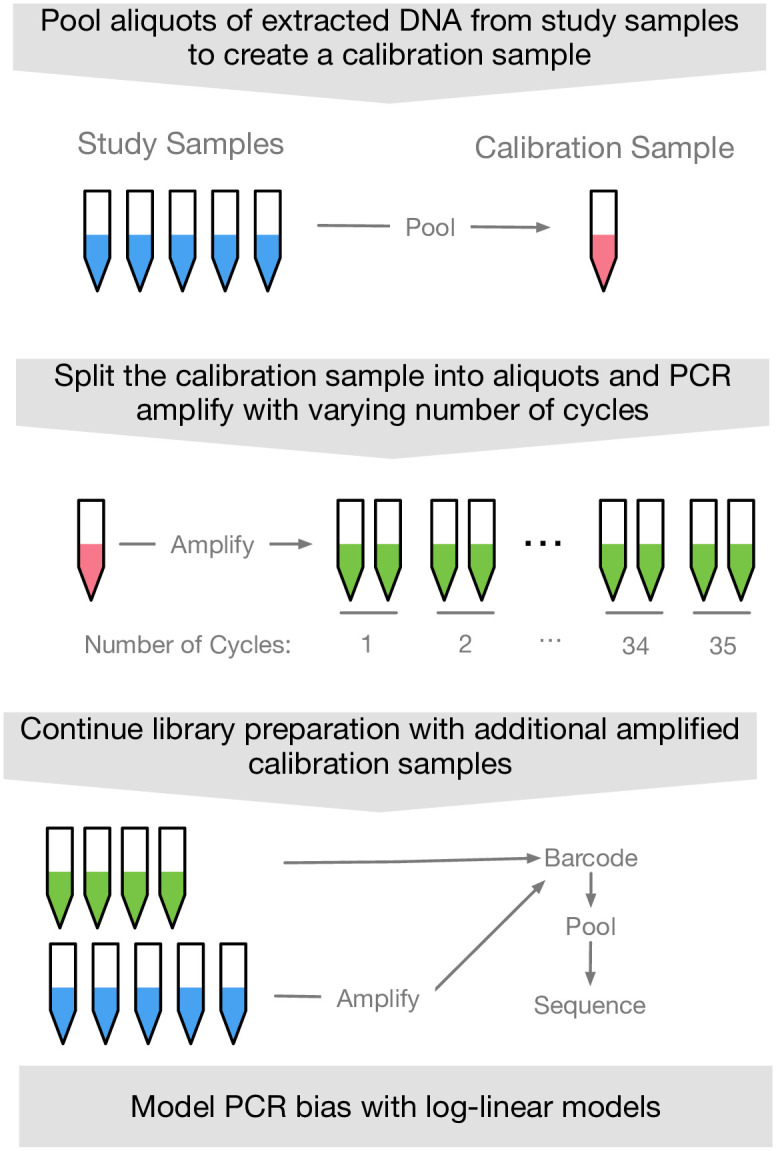
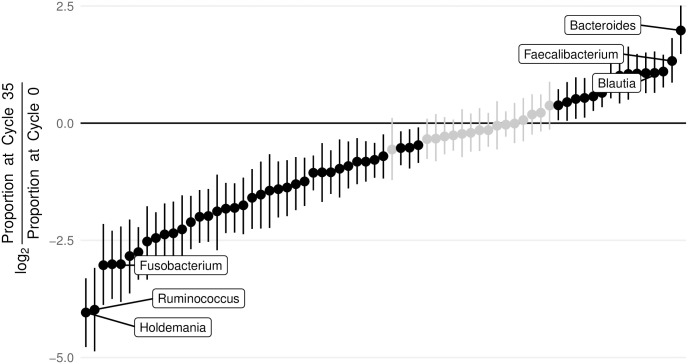
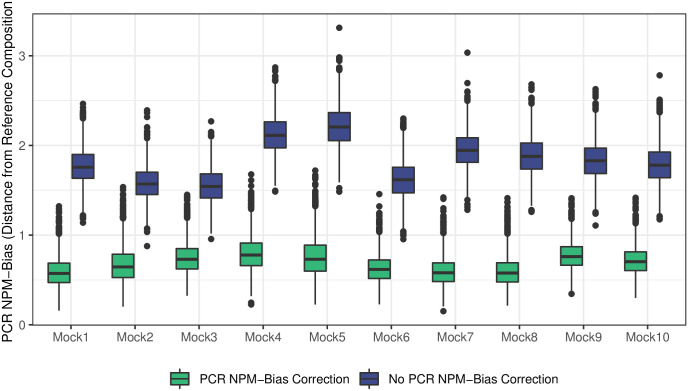
PCR amplification plays an integral role in the measurement of mixed microbial communities via high-throughput DNA sequencing of the 16S ribosomal RNA (rRNA) gene. Yet PCR is also known to introduce multiple forms of bias in 16S rRNA studies. Here we present a paired modeling and experimental approach to characterize and mitigate PCR NPM-bias (PCR bias from non-primer-mismatch sources) in microbiota surveys. We use experimental data from mock bacterial communities to validate our approach and human gut microbiota samples to characterize PCR NPM-bias under real-world conditions. Our results suggest that PCR NPM-bias can skew estimates of microbial relative abundances by a factor of 4 or more, but that this bias can be mitigated using log-ratio linear models.
PCR 扩增在通过高通量 DNA 测序对 16S 核糖体 RNA(rRNA) 基因进行混合微生物群落的测量中起着重要作用。然而,PCR 也已知会在 16S rRNA 研究中引入多种形式的偏差。在这里,我们提出了一种配对的建模和实验方法来描述和减轻微生物组调查中 PCR 非配对错配偏倚(来自非引物错配源的 PCR 偏倚)。我们使用来自模拟细菌群落的实验数据来验证我们的方法,并使用人类肠道微生物组样本在实际条件下描述 PCR 非配对错配偏倚。我们的结果表明,PCR 非配对错配偏倚可能会使微生物相对丰度的估计值偏差 4 倍或更多,但可以使用对数比线性模型来减轻这种偏倚。