Plant Genetics Laboratory, Embrapa-Recursos Genéticos e Biotecnologia, Parque Estação Biológica, Brasília 70770-970, DF, Brazil.
BMC Genomics. 2011 Apr 14;12:189. doi: 10.1186/1471-2164-12-189.
Technological advances are progressively increasing the application of genomics to a wider array of economically and ecologically important species. High-density maps enriched for transcribed genes facilitate the discovery of connections between genes and phenotypes. We report the construction of a high-density linkage map of expressed genes for the heterozygous genome of Eucalyptus using Single Feature Polymorphism (SFP) markers.
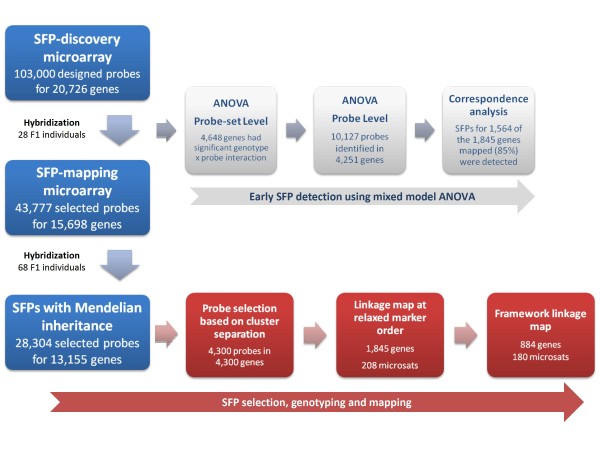
SFP discovery and mapping was achieved using pseudo-testcross screening and selective mapping to simultaneously optimize linkage mapping and microarray costs. SFP genotyping was carried out by hybridizing complementary RNA prepared from 4.5 year-old trees xylem to an SFP array containing 103,000 25-mer oligonucleotide probes representing 20,726 unigenes derived from a modest size expressed sequence tags collection. An SFP-mapping microarray with 43,777 selected candidate SFP probes representing 15,698 genes was subsequently designed and used to genotype SFPs in a larger subset of the segregating population drawn by selective mapping. A total of 1,845 genes were mapped, with 884 of them ordered with high likelihood support on a framework map anchored to 180 microsatellites with average density of 1.2 cM. Using more probes per unigene increased by two-fold the likelihood of detecting segregating SFPs eventually resulting in more genes mapped. In silico validation showed that 87% of the SFPs map to the expected location on the 4.5X draft sequence of the Eucalyptus grandis genome.
The Eucalyptus 1,845 gene map is the most highly enriched map for transcriptional information for any forest tree species to date. It represents a major improvement on the number of genes previously positioned on Eucalyptus maps and provides an initial glimpse at the gene space for this global tree genome. A general protocol is proposed to build high-density transcript linkage maps in less characterized plant species by SFP genotyping with a concurrent objective of reducing microarray costs. HIgh-density gene-rich maps represent a powerful resource to assist gene discovery endeavors when used in combination with QTL and association mapping and should be especially valuable to assist the assembly of reference genome sequences soon to come for several plant and animal species.
技术进步正在逐渐将基因组学应用于更广泛的具有经济和生态重要性的物种。高密度图谱富集转录基因,有助于发现基因与表型之间的联系。我们报告了利用单特征多态性(SFP)标记构建杂种基因组桉树表达基因高密度连锁图谱的方法。
通过假测交筛选和选择性作图同时优化连锁作图和微阵列成本,实现了 SFP 的发现和作图。通过杂交来自 4.5 年生树木木质部的互补 RNA 与包含 20726 个来自适度大小表达序列标签集合的单基因的 103000 个 25 碱基寡核苷酸探针的 SFP 阵列进行 SFP 基因型分析。随后设计并使用包含 15698 个基因的 43777 个选定候选 SFP 探针的 SFP 作图微阵列对通过选择性作图选择的分离群体中的更大亚群进行 SFP 基因分型。共定位了 1845 个基因,其中 884 个基因以高可信度排序在一个基于 180 个微卫星的框架图谱上,平均密度为 1.2cM。每个单基因使用更多的探针可使检测到分离 SFP 的可能性增加一倍,最终可定位更多的基因。通过对计算机进行验证,87%的 SFP 映射到拟南芥 4.5X 草图序列的预期位置。
桉树 1845 个基因图谱是迄今为止转录信息最丰富的森林树种图谱。它比以前在桉树图谱上定位的基因数量有了显著提高,并为这个全球树种的基因空间提供了初步的了解。通过 SFP 基因分型,同时降低微阵列成本,提出了在特征不明显的植物物种中构建高密度转录连锁图谱的一般方案。高密度基因丰富图谱是一个强大的资源,有助于基因发现工作,当与 QTL 和关联作图结合使用时,应该特别有助于组装即将推出的几个植物和动物物种的参考基因组序列。