School of Plant Science and CRC for Forestry, University of Tasmania, Private Bag 55 Hobart, Tasmania, 7001, Australia.
BMC Genomics. 2012 Jun 15;13:240. doi: 10.1186/1471-2164-13-240.
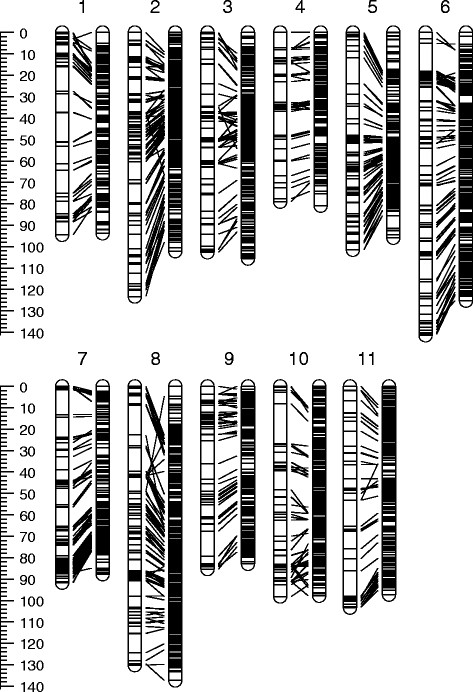
Genetic linkage maps are invaluable resources in plant research. They provide a key tool for many genetic applications including: mapping quantitative trait loci (QTL); comparative mapping; identifying unlinked (i.e. independent) DNA markers for fingerprinting, population genetics and phylogenetics; assisting genome sequence assembly; relating physical and recombination distances along the genome and map-based cloning of genes. Eucalypts are the dominant tree species in most Australian ecosystems and of economic importance globally as plantation trees. The genome sequence of E. grandis has recently been released providing unprecedented opportunities for genetic and genomic research in the genus. A robust reference linkage map containing sequence-based molecular markers is needed to capitalise on this resource. Several high density linkage maps have recently been constructed for the main commercial forestry species in the genus (E. grandis, E. urophylla and E. globulus) using sequenced Diversity Arrays Technology (DArT) and microsatellite markers. To provide a single reference linkage map for eucalypts a composite map was produced through the integration of data from seven independent mapping experiments (1950 individuals) using a marker-merging method.
The composite map totalled 1107 cM and contained 4101 markers; comprising 3880 DArT, 213 microsatellite and eight candidate genes. Eighty-one DArT markers were mapped to two or more linkage groups, resulting in the 4101 markers being mapped to 4191 map positions. Approximately 13% of DArT markers mapped to identical map positions, thus the composite map contained 3634 unique loci at an average interval of 0.31 cM.
The composite map represents the most saturated linkage map yet produced in Eucalyptus. As the majority of DArT markers contained on the map have been sequenced, the map provides a direct link to the E. grandis genome sequence and will serve as an important reference for progressing eucalypt research.
遗传连锁图谱在植物研究中是非常宝贵的资源。它们为许多遗传应用提供了关键工具,包括:定位数量性状基因座(QTL);比较作图;为指纹图谱、群体遗传学和系统发育学鉴定不连锁(即独立)的 DNA 标记;协助基因组序列组装;关联基因组上的物理和重组距离;以及基于图谱的基因克隆。桉树是大多数澳大利亚生态系统中的主要树种,也是全球经济中重要的造林树种。大果桉的基因组序列最近已经公布,这为该属的遗传和基因组研究提供了前所未有的机会。需要构建一个稳健的参考连锁图谱,其中包含基于序列的分子标记,以充分利用这一资源。最近,已经使用测序多样性阵列技术(DArT)和微卫星标记构建了几个主要商业造林树种(大果桉、尾巨桉和蓝桉)的高密度连锁图谱。为了提供桉树的单一参考连锁图谱,通过整合来自七个独立作图实验(1950 个个体)的数据,使用标记合并方法,制作了一个复合图谱。
该复合图谱总长 1107cm,包含 4101 个标记,包括 3880 个 DArT、213 个微卫星和 8 个候选基因。81 个 DArT 标记映射到两个或更多连锁群,导致 4101 个标记映射到 4191 个图谱位置。大约 13%的 DArT 标记映射到相同的图谱位置,因此复合图谱包含 3634 个独特的位点,平均间隔为 0.31cm。
该复合图谱代表了迄今在桉树中产生的最饱和的连锁图谱。由于图谱上的大多数 DArT 标记都已测序,因此该图谱直接与大果桉基因组序列相关联,将成为推进桉树研究的重要参考。