University of Washington, Department of Biochemistry and HHMI, Seattle, Washington 98195, USA.
Nature. 2011 May 26;473(7348):540-3. doi: 10.1038/nature09964. Epub 2011 May 1.
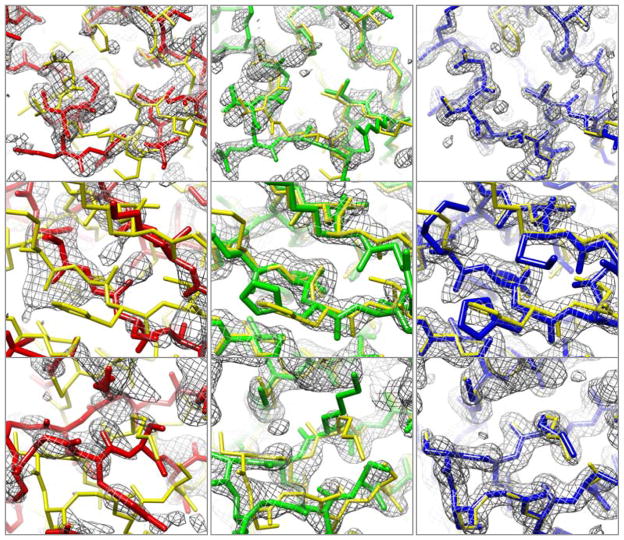
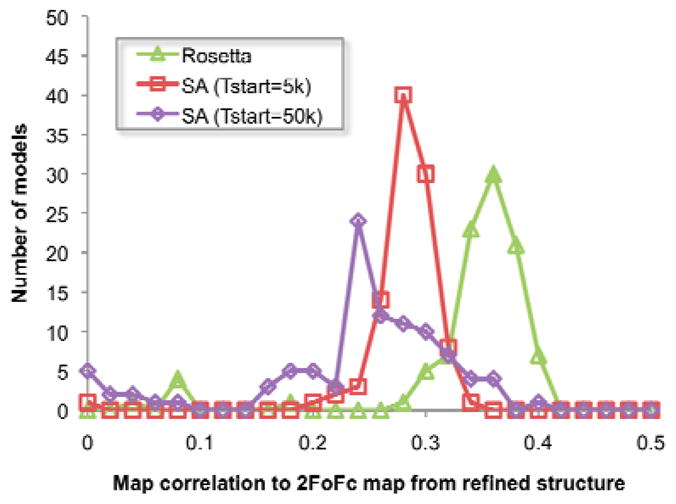
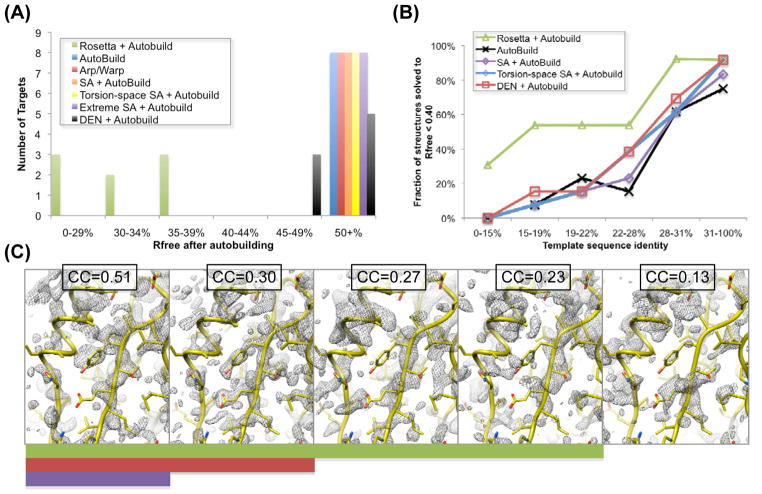
Molecular replacement procedures, which search for placements of a starting model within the crystallographic unit cell that best account for the measured diffraction amplitudes, followed by automatic chain tracing methods, have allowed the rapid solution of large numbers of protein crystal structures. Despite extensive work, molecular replacement or the subsequent rebuilding usually fail with more divergent starting models based on remote homologues with less than 30% sequence identity. Here we show that this limitation can be substantially reduced by combining algorithms for protein structure modelling with those developed for crystallographic structure determination. An approach integrating Rosetta structure modelling with Autobuild chain tracing yielded high-resolution structures for 8 of 13 X-ray diffraction data sets that could not be solved in the laboratories of expert crystallographers and that remained unsolved after application of an extensive array of alternative approaches. We estimate that the new method should allow rapid structure determination without experimental phase information for over half the cases where current methods fail, given diffraction data sets of better than 3.2 Å resolution, four or fewer copies in the asymmetric unit, and the availability of structures of homologous proteins with >20% sequence identity.
分子置换程序通过在晶体学单位晶胞中搜索最佳的起始模型位置来解释测量的衍射强度,随后使用自动链追踪方法,可以快速解决大量蛋白质晶体结构的问题。尽管已经进行了广泛的研究,但分子置换或随后的重建通常会失败,尤其是对于基于与序列相似性小于 30%的远缘同源物的起始模型。在这里,我们表明,通过将蛋白质结构建模算法与晶体学结构确定算法相结合,可以大大减少这种限制。一种将 Rosetta 结构建模与 Autobuild 链追踪相结合的方法,为 13 个 X 射线衍射数据集的 8 个数据集提供了高分辨率结构,这些数据集无法在专家晶体学家的实验室中解决,并且在应用了广泛的替代方法后仍然未解决。我们估计,在目前的方法失败的情况下,对于超过一半的情况,新方法应该能够在没有实验相位信息的情况下快速确定结构,前提是衍射数据集的分辨率优于 3.2Å,不对称单元中只有四个或更少的副本,并且具有>20%序列相似性的同源蛋白结构可用。