Institute of Molecular Biology, University of Oregon, Eugene, Oregon, United States of America.
PLoS One. 2011 Apr 13;6(4):e18561. doi: 10.1371/journal.pone.0018561.
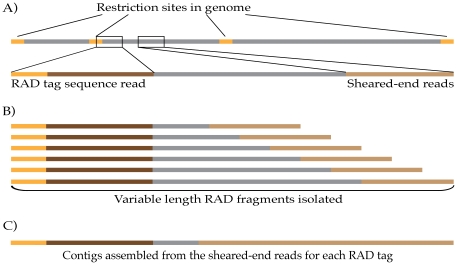
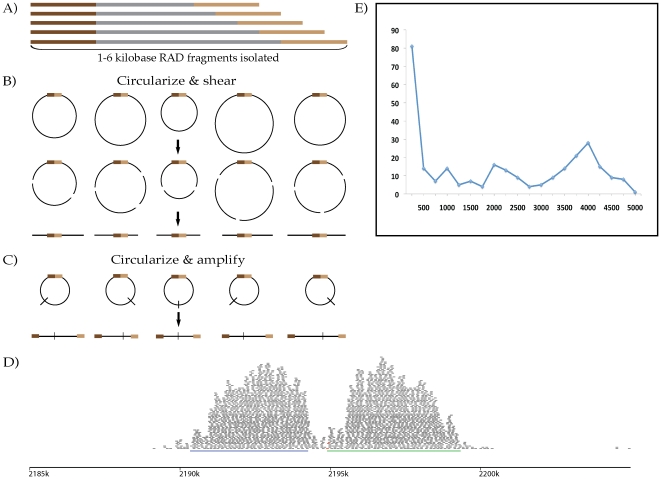
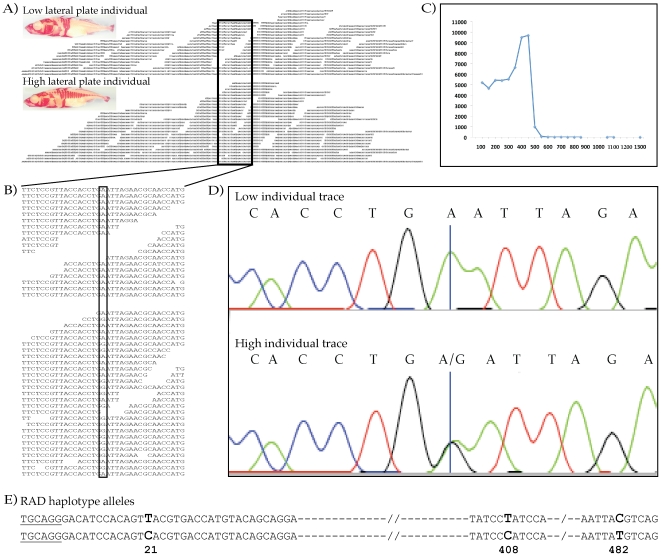
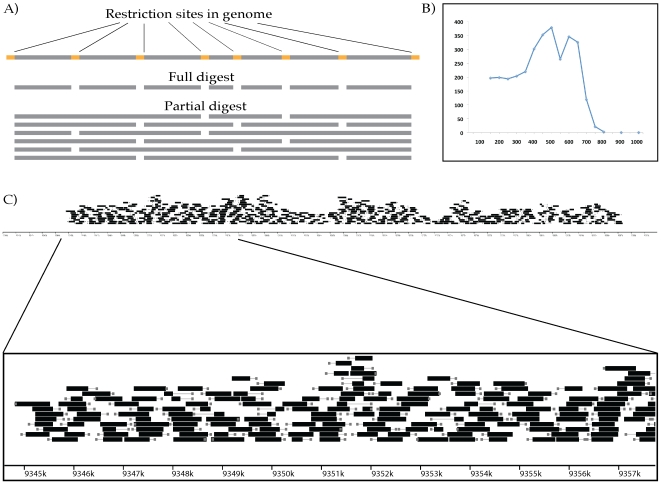
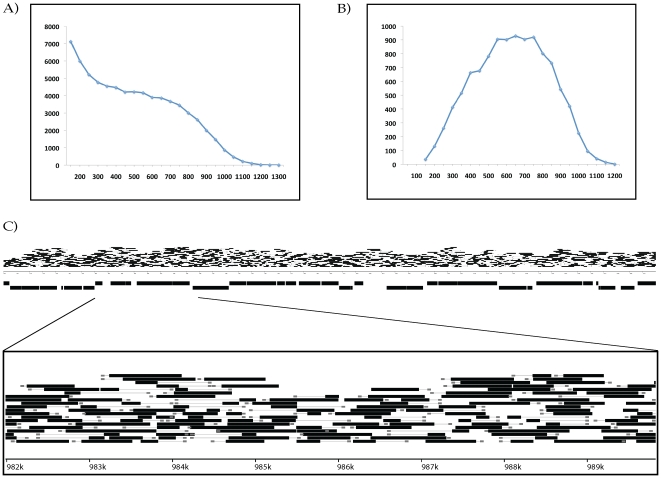
Despite the power of massively parallel sequencing platforms, a drawback is the short length of the sequence reads produced. We demonstrate that short reads can be locally assembled into longer contigs using paired-end sequencing of restriction-site associated DNA (RAD-PE) fragments. We use this RAD-PE contig approach to identify single nucleotide polymorphisms (SNPs) and determine haplotype structure in threespine stickleback and to sequence E. coli and stickleback genomic DNA with overlapping contigs of several hundred nucleotides. We also demonstrate that adding a circularization step allows the local assembly of contigs up to 5 kilobases (kb) in length. The ease of assembly and accuracy of the individual contigs produced from each RAD site sequence suggests RAD-PE sequencing is a useful way to convert genome-wide short reads into individually-assembled sequences hundreds or thousands of nucleotides long.
尽管大规模并行测序平台功能强大,但也存在一个缺点,即生成的序列读长较短。我们证明,通过对限制性位点相关 DNA(RAD-PE)片段进行成对测序,可以将短读段局部组装成长片段。我们使用这种 RAD-PE 片段组装方法来鉴定三刺鱼中的单核苷酸多态性(SNP)和单倍型结构,并对大肠杆菌和三刺鱼基因组 DNA 进行测序,获得了数百个核苷酸重叠的重叠片段。我们还证明,添加环化步骤可以允许将长度达 5 千碱基(kb)的片段进行局部组装。每个 RAD 位点序列产生的组装片段的易组装性和准确性表明,RAD-PE 测序是一种将全基因组短读段转化为数百或数千个核苷酸长的单独组装序列的有效方法。