Institute for Hygiene and Microbiology, University of Würzburg, Würzburg, Germany.
PLoS One. 2011 Apr 26;6(4):e18441. doi: 10.1371/journal.pone.0018441.
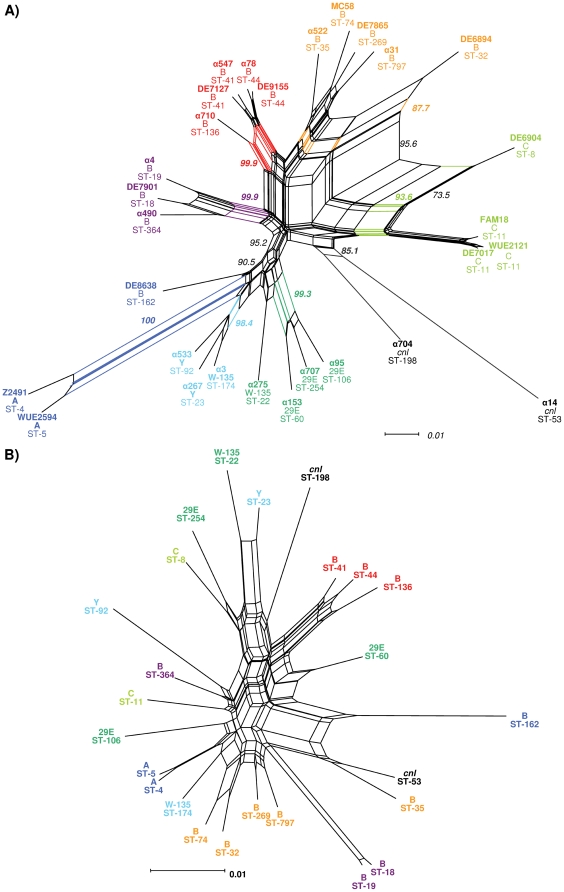
Neisseria meningitidis is a naturally transformable, facultative pathogen colonizing the human nasopharynx. Here, we analyze on a genome-wide level the impact of recombination on gene-complement diversity and virulence evolution in N. meningitidis. We combined comparative genome hybridization using microarrays (mCGH) and multilocus sequence typing (MLST) of 29 meningococcal isolates with computational comparison of a subset of seven meningococcal genome sequences.
We found that lateral gene transfer of minimal mobile elements as well as prophages are major forces shaping meningococcal population structure. Extensive gene content comparison revealed novel associations of virulence with genetic elements besides the recently discovered meningococcal disease associated (MDA) island. In particular, we identified an association of virulence with a recently described canonical genomic island termed IHT-E and a differential distribution of genes encoding RTX toxin- and two-partner secretion systems among hyperinvasive and non-hyperinvasive lineages. By computationally screening also the core genome for signs of recombination, we provided evidence that about 40% of the meningococcal core genes are affected by recombination primarily within metabolic genes as well as genes involved in DNA replication and repair. By comparison with the results of previous mCGH studies, our data indicated that genetic structuring as revealed by mCGH is stable over time and highly similar for isolates from different geographic origins.
Recombination comprising lateral transfer of entire genes as well as homologous intragenic recombination has a profound impact on meningococcal population structure and genome composition. Our data support the hypothesis that meningococcal virulence is polygenic in nature and that differences in metabolism might contribute to virulence.
脑膜炎奈瑟菌是一种可自然转化的兼性病原体,定植于人体鼻咽部。在此,我们在全基因组水平上分析了重组对脑膜炎奈瑟菌基因补体多样性和毒力进化的影响。我们结合了使用微阵列的比较基因组杂交 (mCGH) 和 29 株脑膜炎奈瑟菌的多位点序列分型 (MLST),并对 7 株脑膜炎奈瑟菌基因组序列的子集进行了计算比较。
我们发现,最小移动元件和原噬菌体的水平基因转移是塑造脑膜炎奈瑟菌群体结构的主要力量。广泛的基因内容比较揭示了除最近发现的脑膜炎球菌病相关 (MDA) 岛之外,毒力与遗传元件之间存在新的关联。特别是,我们发现与最近描述的称为 IHT-E 的经典基因组岛以及编码 RTX 毒素和双组分分泌系统的基因在高侵袭性和非高侵袭性谱系中的差异分布有关。通过计算性地筛选核心基因组中的重组迹象,我们提供了证据表明,大约 40%的脑膜炎奈瑟菌核心基因受到重组的影响,主要是在代谢基因以及参与 DNA 复制和修复的基因中。与之前 mCGH 研究的结果相比,我们的数据表明,mCGH 揭示的遗传结构在时间上是稳定的,并且对于来自不同地理起源的分离株高度相似。
包括整个基因的侧向转移以及同源基因内重组的重组对脑膜炎奈瑟菌的群体结构和基因组组成有深远的影响。我们的数据支持这样的假设,即脑膜炎奈瑟菌的毒力本质上是多基因的,并且代谢差异可能有助于毒力。