Cancer Research UK Centre, Leeds Institute of Molecular Medicine, St. James's University Hospital, Leeds, UK.
Proteomics. 2011 Jun;11(11):2222-35. doi: 10.1002/pmic.201100005. Epub 2011 May 5.
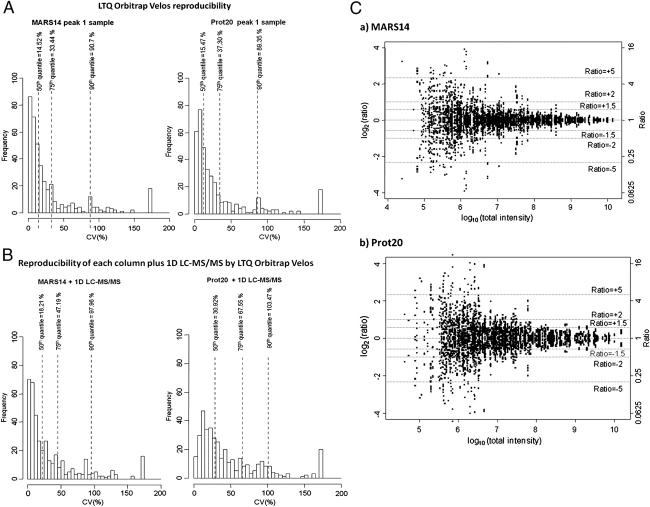
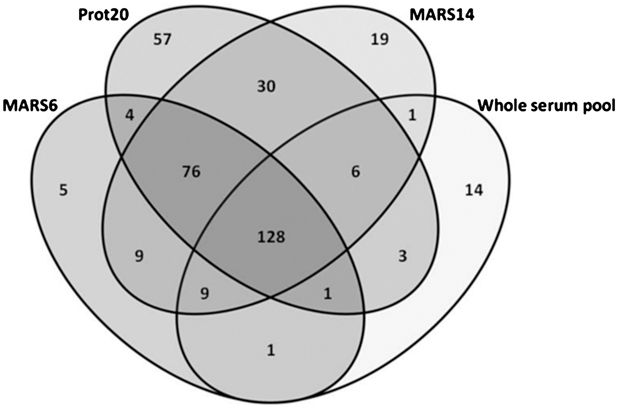
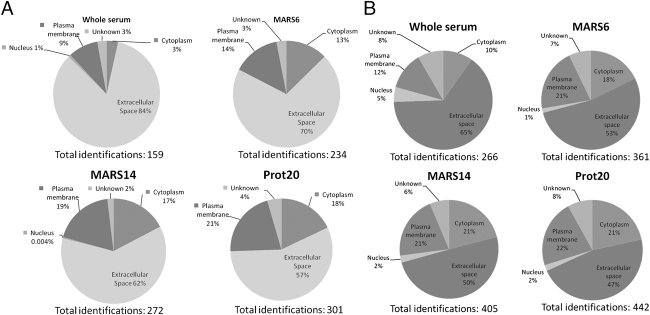
Immunodepletion of clinical fluids to overcome the dominance by a few very abundant proteins has been explored but studies are few, commonly examining only limited aspects with one analytical platform. We have systematically compared immunodepletion of 6, 14, or 20 proteins using serum from renal transplant patients, analysing reproducibility, depth of coverage, efficiency, and specificity using 2-D DIGE ('top-down') and LC-MS/MS ('bottom-up'). A progressive increase in protein number (≥2 unique peptides) was found from 159 in unfractionated serum to 301 following 20 protein depletion using a relatively high-throughput 1-D-LC-MS/MS approach, including known biomarkers and moderate-lower abundance proteins such as NGAL and cytokine/growth factor receptors. On the contrary, readout by 2-D DIGE demonstrated good reproducibility of immunodepletion, but additional proteins seen tended to be isoforms of existing proteins. Depletion of 14 or 20 proteins followed by LC-MS/MS showed excellent reproducibility of proteins detected and a significant overlap between columns. Using label-free analysis, greater run-to-run variability was seen with the Prot20 column compared with the MARS14 column (median %CVs of 30.9 versus 18.2%, respectively) and a corresponding wider precision profile for the Prot20. These results illustrate the potential of immunodepletion followed by 1-D nano-LC-LTQ Orbitrap Velos analysis in a moderate through-put biomarker discovery process.
免疫耗尽临床液体以克服少数非常丰富的蛋白质的优势已经被探索过,但研究很少,通常只使用一种分析平台来检查有限的方面。我们系统地比较了使用肾移植患者的血清进行的 6、14 或 20 种蛋白质的免疫耗尽,使用 2-D DIGE(“自上而下”)和 LC-MS/MS(“自下而上”)分析重复性、覆盖率深度、效率和特异性。在使用相对高通量的 1-D-LC-MS/MS 方法(包括已知生物标志物和中低丰度蛋白质如 NGAL 和细胞因子/生长因子受体)对 20 种蛋白质进行耗尽后,从未分馏血清中的 159 种蛋白质增加到 301 种蛋白质,发现蛋白质数量(≥2 个独特肽)呈递增趋势。相反,2-D DIGE 的读出显示免疫耗尽的良好重现性,但观察到的额外蛋白质往往是现有蛋白质的同工型。通过 LC-MS/MS 耗尽 14 或 20 种蛋白质后,检测到的蛋白质具有极好的重现性,并且列之间存在显著重叠。使用无标记分析,与 MARS14 柱相比,Prot20 柱的运行到运行变异性更大(中位数 %CVs 分别为 30.9%和 18.2%),Prot20 柱的精度分布也相应变宽。这些结果说明了在中等通量生物标志物发现过程中,免疫耗尽后进行 1-D 纳米 LC-LTQ Orbitrap Velos 分析的潜力。