Institute for Evolution and Biodiversity, Westfalian Wilhelms University, Hüfferstrasse 1, 48149 Münster, Germany.
BMC Genomics. 2011 May 11;12:227. doi: 10.1186/1471-2164-12-227.
The garden pea, Pisum sativum, is among the best-investigated legume plants and of significant agro-commercial relevance. Pisum sativum has a large and complex genome and accordingly few comprehensive genomic resources exist.
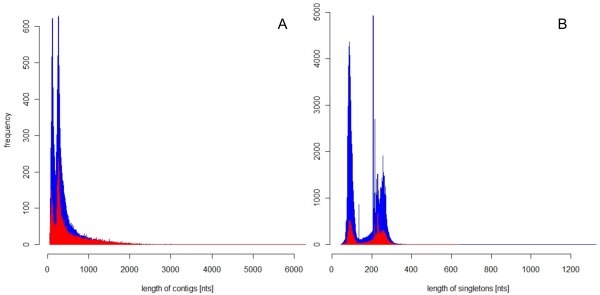
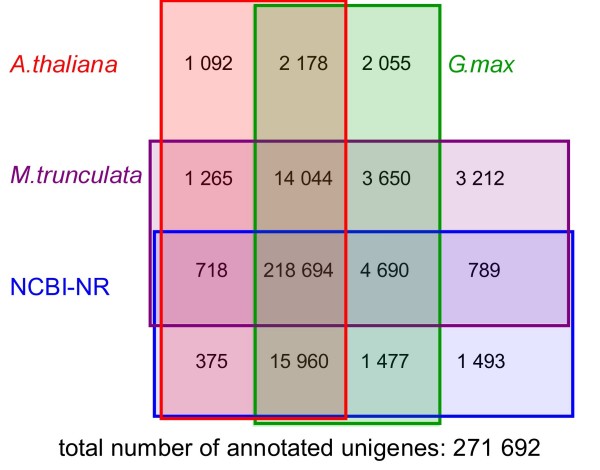
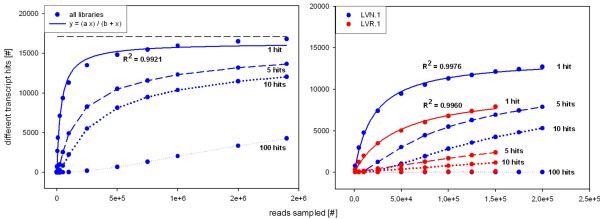
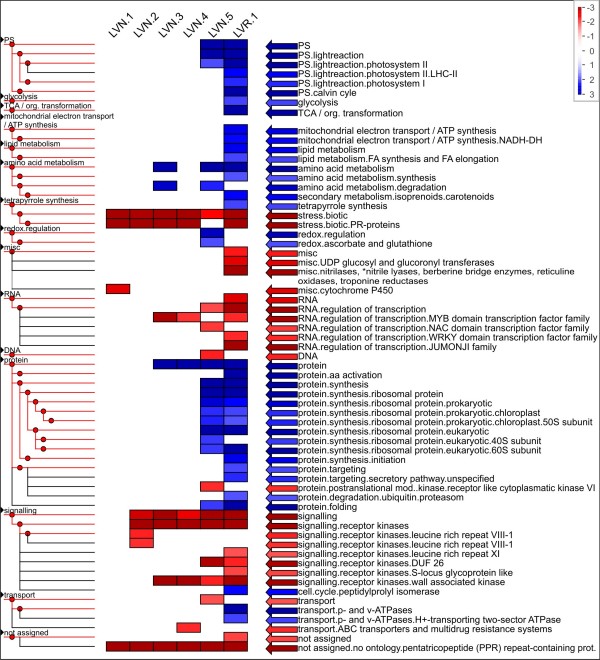
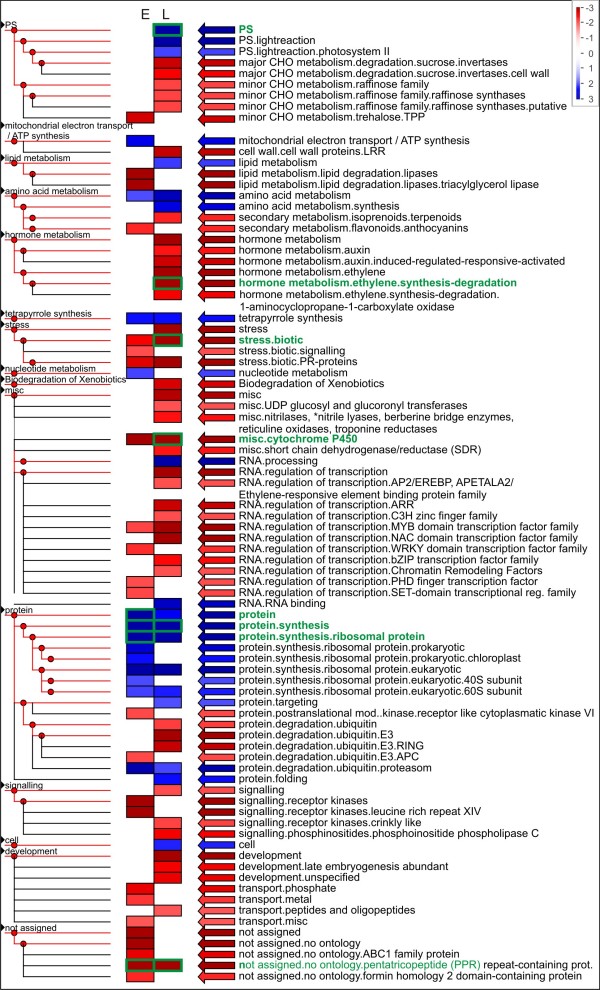
We analyzed the pea transcriptome at the highest possible amount of accuracy by current technology. We used next generation sequencing with the Roche/454 platform and evaluated and compared a variety of approaches, including diverse tissue libraries, normalization, alternative sequencing technologies, saturation estimation and diverse assembly strategies. We generated libraries from flowers, leaves, cotyledons, epi- and hypocotyl, and etiolated and light treated etiolated seedlings, comprising a total of 450 megabases. Libraries were assembled into 324,428 unigenes in a first pass assembly.A second pass assembly reduced the amount to 81,449 unigenes but caused a significant number of chimeras. Analyses of the assemblies identified the assembly step as a major possibility for improvement. By recording frequencies of Arabidopsis orthologs hit by randomly drawn reads and fitting parameters of the saturation curve we concluded that sequencing was exhaustive. For leaf libraries we found normalization allows partial recovery of expression strength aside the desired effect of increased coverage. Based on theoretical and biological considerations we concluded that the sequence reads in the database tagged the vast majority of transcripts in the aerial tissues. A pathway representation analysis showed the merits of sampling multiple aerial tissues to increase the number of tagged genes. All results have been made available as a fully annotated database in fasta format.
We conclude that the approach taken resulted in a high quality - dataset which serves well as a first comprehensive reference set for the model legume pea. We suggest future deep sequencing transcriptome projects of species lacking a genomics backbone will need to concentrate mainly on resolving the issues of redundancy and paralogy during transcriptome assembly.
花园豌豆(Pisum sativum)是研究最深入的豆科植物之一,具有重要的农业商业意义。Pisum sativum 具有庞大而复杂的基因组,因此很少有全面的基因组资源。
我们利用罗氏/454 平台的下一代测序技术,以目前最高的精度分析了豌豆转录组。我们评估和比较了多种方法,包括不同的组织文库、归一化、替代测序技术、饱和度估计和不同的组装策略。我们从花、叶、子叶、上胚轴和下胚轴以及黄化和光处理的黄化苗中生成文库,总共产生了 450 兆碱基的文库。在初步组装中,文库被组装成 324428 个基因。第二次组装将数量减少到 81449 个基因,但产生了大量嵌合体。组装分析表明,组装步骤是改进的主要可能性。通过记录随机读取的拟南芥直系同源物的频率并拟合饱和度曲线的参数,我们得出结论,测序是详尽的。对于叶文库,我们发现归一化除了增加覆盖范围的预期效果外,还允许部分恢复表达强度。基于理论和生物学考虑,我们得出结论,数据库中的序列读取标记了空中组织中绝大多数的转录本。途径表示分析表明,从多个空中组织中采样以增加标记基因的数量是可取的。所有结果均以快速格式的完全注释数据库形式提供。
我们得出的结论是,所采用的方法产生了高质量的数据集,可作为模式豆科植物豌豆的第一个全面参考数据集。我们建议未来缺乏基因组学基础的物种的深度测序转录组项目将需要主要集中解决转录组组装过程中的冗余和同源问题。