Genome Informatics Group, Faculty of Technology and Institute for Bioinformatics, Center for Biotechnology, Bielefeld University, Bielefeld, Germany.
Nucleic Acids Res. 2011 Aug;39(14):e91. doi: 10.1093/nar/gkr225. Epub 2011 May 17.
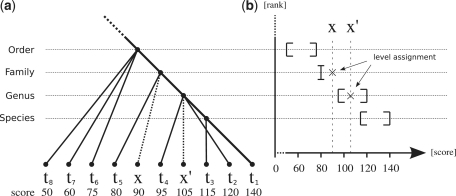
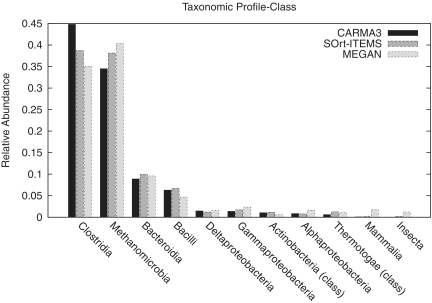
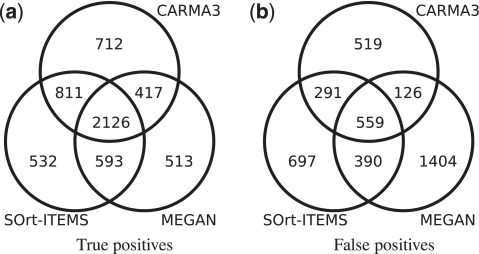
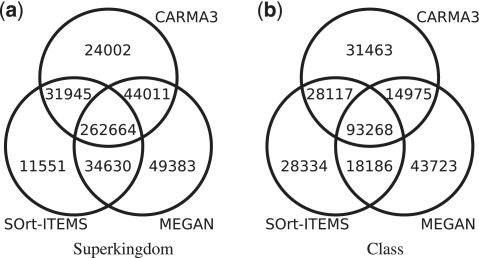
The vast majority of microbes are unculturable and thus cannot be sequenced by means of traditional methods. High-throughput sequencing techniques like 454 or Solexa-Illumina make it possible to explore those microbes by studying whole natural microbial communities and analysing their biological diversity as well as the underlying metabolic pathways. Over the past few years, different methods have been developed for the taxonomic and functional characterization of metagenomic shotgun sequences. However, the taxonomic classification of metagenomic sequences from novel species without close homologue in the biological sequence databases poses a challenge due to the high number of wrong taxonomic predictions on lower taxonomic ranks. Here we present CARMA3, a new method for the taxonomic classification of assembled and unassembled metagenomic sequences that has been adapted to work with both BLAST and HMMER3 homology searches. We show that our method makes fewer wrong taxonomic predictions (at the same sensitivity) than other BLAST-based methods. CARMA3 is freely accessible via the web application WebCARMA from http://webcarma.cebitec.uni-bielefeld.de.
绝大多数微生物是不可培养的,因此无法通过传统方法进行测序。高通量测序技术(如 454 或 Solexa-Illumina)使得研究整个自然微生物群落并分析其生物多样性以及潜在的代谢途径成为可能。在过去的几年中,已经开发出了多种方法来对宏基因组鸟枪法序列进行分类学和功能描述。然而,由于在生物序列数据库中没有密切同源物的新型物种的宏基因组序列的分类学分类具有挑战性,因为在较低的分类等级上存在大量错误的分类预测。在这里,我们提出了 CARMA3,这是一种针对组装和未组装的宏基因组序列的分类学分类的新方法,已经适应了 BLAST 和 HMMER3 同源搜索的使用。我们表明,我们的方法比其他基于 BLAST 的方法做出的错误分类预测更少(在相同的灵敏度下)。CARMA3 可通过 Web 应用程序 WebCARMA 从 http://webcarma.cebitec.uni-bielefeld.de 免费访问。