Department of Medicinal Chemistry, University of Vienna, Vienna, Austria.
PLoS Comput Biol. 2011 May;7(5):e1002036. doi: 10.1371/journal.pcbi.1002036. Epub 2011 May 12.
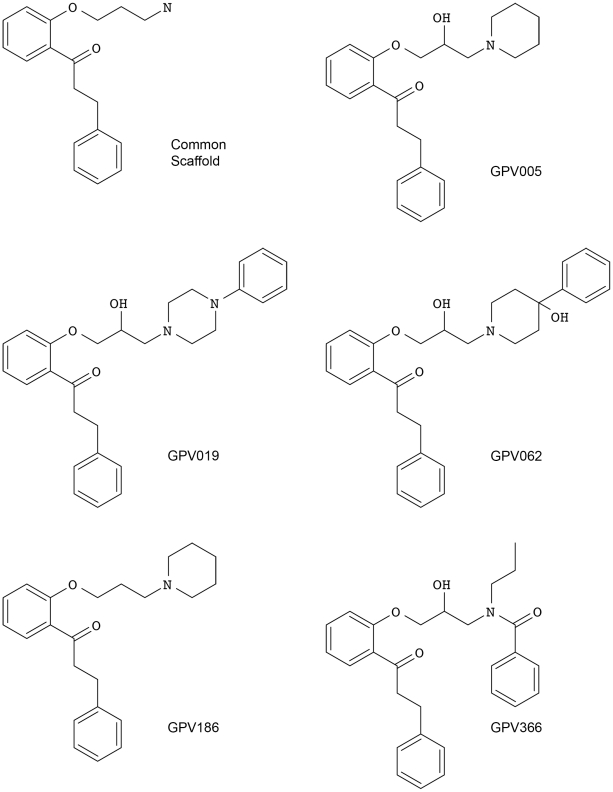
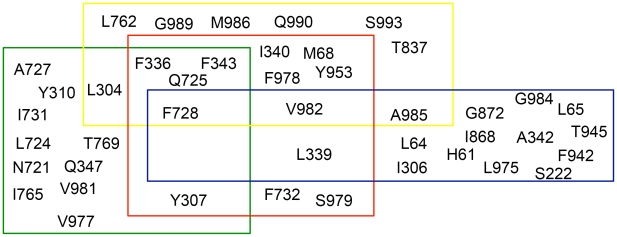
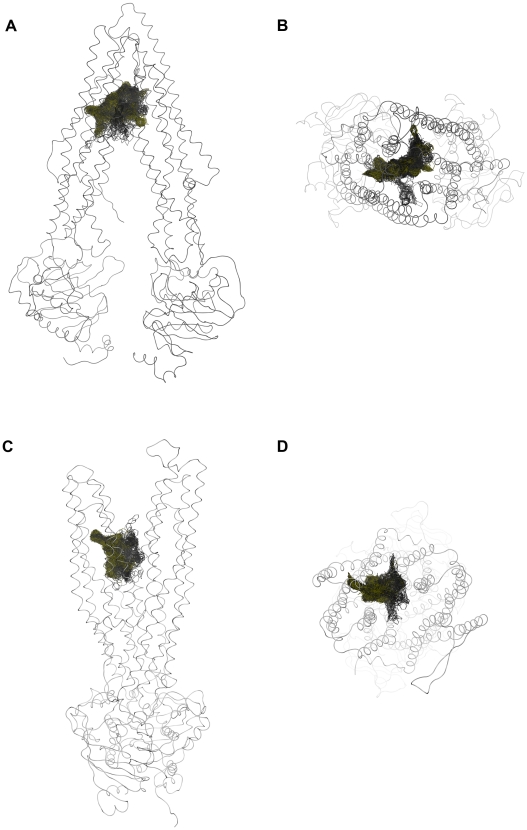
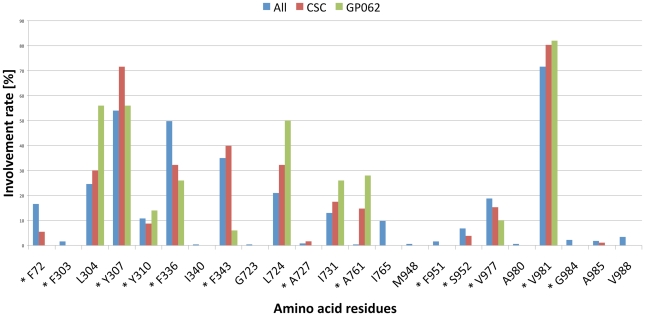
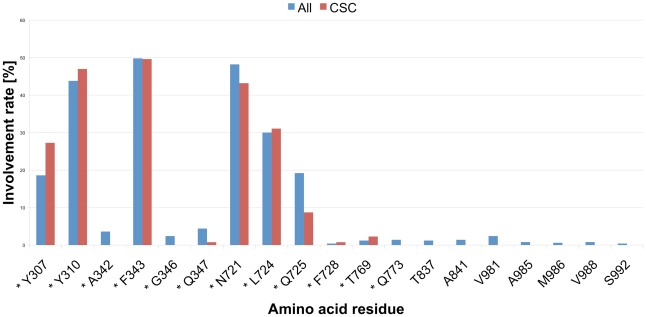
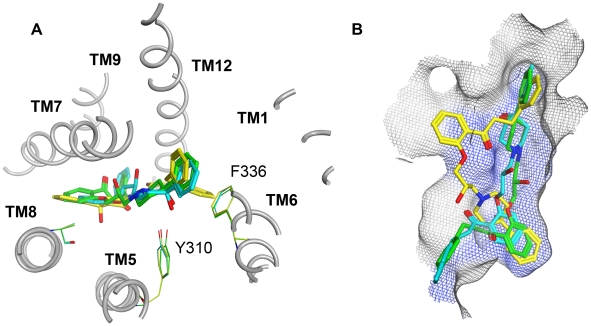
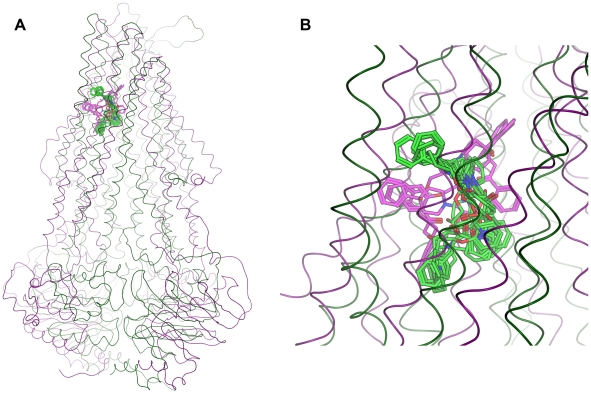
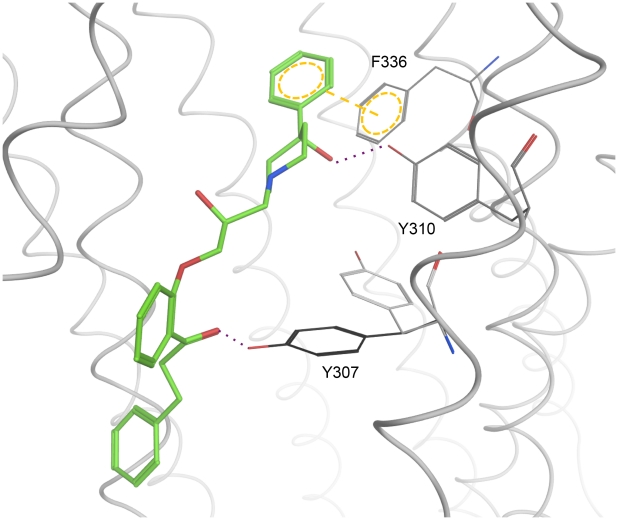
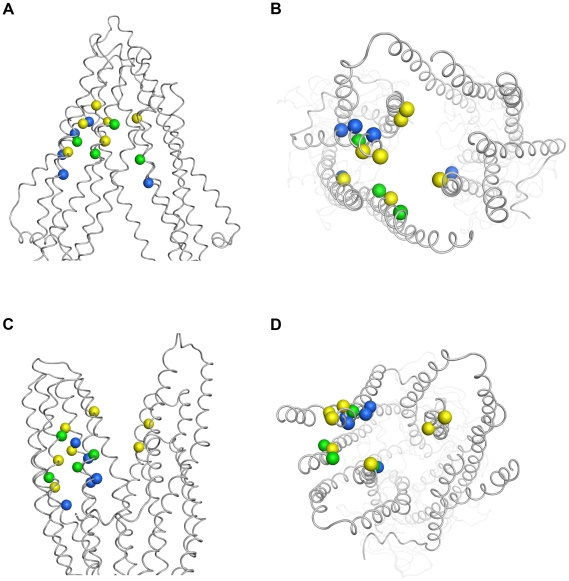
Overexpression of the xenotoxin transporter P-glycoprotein (P-gp) represents one major reason for the development of multidrug resistance (MDR), leading to the failure of antibiotic and cancer therapies. Inhibitors of P-gp have thus been advocated as promising candidates for overcoming the problem of MDR. However, due to lack of a high-resolution structure the concrete mode of interaction of both substrates and inhibitors is still not known. Therefore, structure-based design studies have to rely on protein homology models. In order to identify binding hypotheses for propafenone-type P-gp inhibitors, five different propafenone derivatives with known structure-activity relationship (SAR) pattern were docked into homology models of the apo and the nucleotide-bound conformation of the transporter. To circumvent the uncertainty of scoring functions, we exhaustively sampled the pose space and analyzed the poses by combining information retrieved from SAR studies with common scaffold clustering. The results suggest propafenone binding at the transmembrane helices 5, 6, 7 and 8 in both models, with the amino acid residue Y307 playing a crucial role. The identified binding site in the non-energized state is overlapping with, but not identical to, known binding areas of cyclic P-gp inhibitors and verapamil. These findings support the idea of several small binding sites forming one large binding cavity. Furthermore, the binding hypotheses for both catalytic states were analyzed and showed only small differences in their protein-ligand interaction fingerprints, which indicates only small movements of the ligand during the catalytic cycle.
外排泵 P-糖蛋白(P-gp)的过度表达是多药耐药(MDR)发展的主要原因之一,导致抗生素和癌症治疗失败。因此,P-gp 的抑制剂被认为是克服 MDR 问题的有希望的候选药物。然而,由于缺乏高分辨率结构,仍然不清楚底物和抑制剂的具体相互作用模式。因此,基于结构的设计研究必须依赖于蛋白质同源模型。为了确定普罗帕酮型 P-gp 抑制剂的结合假说,将五种具有已知结构-活性关系(SAR)模式的不同普罗帕酮衍生物对接入到转运蛋白的apo 和核苷酸结合构象的同源模型中。为了避免评分函数的不确定性,我们详尽地采样了构象空间,并通过将 SAR 研究中检索到的信息与常见支架聚类相结合来分析构象。结果表明,在两种模型中,普罗帕酮结合在跨膜螺旋 5、6、7 和 8 上,其中氨基酸残基 Y307 起着关键作用。在非能量状态下确定的结合位点与环状 P-gp 抑制剂和维拉帕米的已知结合区域重叠,但不完全相同。这些发现支持了几个小的结合位点形成一个大的结合腔的想法。此外,还分析了两种催化状态的结合假说,发现它们的蛋白-配体相互作用指纹之间只有很小的差异,这表明在催化循环中配体只有很小的运动。