Department of Medicine, Stanford University School of Medicine, Stanford, California 94305, USA.
Nature. 2011 May 22;474(7351):399-402. doi: 10.1038/nature10084.
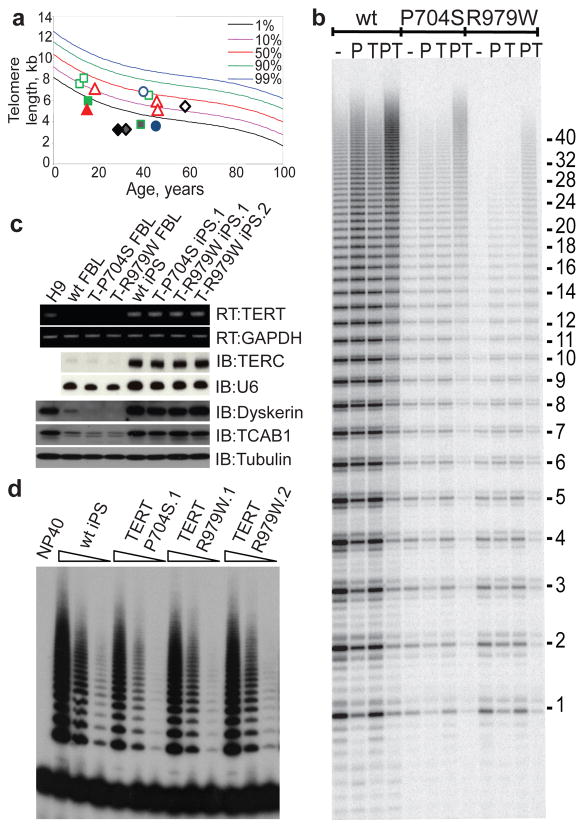
The differentiation of patient-derived induced pluripotent stem cells (iPSCs) to committed fates such as neurons, muscle and liver is a powerful approach for understanding key parameters of human development and disease. Whether undifferentiated iPSCs themselves can be used to probe disease mechanisms is uncertain. Dyskeratosis congenita is characterized by defective maintenance of blood, pulmonary tissue and epidermal tissues and is caused by mutations in genes controlling telomere homeostasis. Short telomeres, a hallmark of dyskeratosis congenita, impair tissue stem cell function in mouse models, indicating that a tissue stem cell defect may underlie the pathophysiology of dyskeratosis congenita. Here we show that even in the undifferentiated state, iPSCs from dyskeratosis congenita patients harbour the precise biochemical defects characteristic of each form of the disease and that the magnitude of the telomere maintenance defect in iPSCs correlates with clinical severity. In iPSCs from patients with heterozygous mutations in TERT, the telomerase reverse transcriptase, a 50% reduction in telomerase levels blunts the natural telomere elongation that accompanies reprogramming. In contrast, mutation of dyskerin (DKC1) in X-linked dyskeratosis congenita severely impairs telomerase activity by blocking telomerase assembly and disrupts telomere elongation during reprogramming. In iPSCs from a form of dyskeratosis congenita caused by mutations in TCAB1 (also known as WRAP53), telomerase catalytic activity is unperturbed, yet the ability of telomerase to lengthen telomeres is abrogated, because telomerase mislocalizes from Cajal bodies to nucleoli within the iPSCs. Extended culture of DKC1-mutant iPSCs leads to progressive telomere shortening and eventual loss of self-renewal, indicating that a similar process occurs in tissue stem cells in dyskeratosis congenita patients. These findings in iPSCs from dyskeratosis congenita patients reveal that undifferentiated iPSCs accurately recapitulate features of a human stem cell disease and may serve as a cell-culture-based system for the development of targeted therapeutics.
将患者来源的诱导多能干细胞(iPSC)分化为神经元、肌肉和肝脏等特化命运是理解人类发育和疾病关键参数的有力方法。未分化的 iPSC 本身是否可用于探究疾病机制尚不确定。先天性角化不良症的特征是血液、肺组织和表皮组织的维持缺陷,由控制端粒动态平衡的基因发生突变引起。端粒较短是先天性角化不良症的标志,在小鼠模型中损害组织干细胞功能,表明组织干细胞缺陷可能是先天性角化不良症的病理生理学基础。在这里,我们表明即使在未分化状态下,来自先天性角化不良症患者的 iPSC 也具有每种疾病形式的精确生化缺陷特征,并且 iPSC 中端粒维持缺陷的严重程度与临床严重程度相关。在 TERT 基因杂合突变的患者 iPSC 中,端粒酶逆转录酶水平降低 50%,会削弱伴随重编程的自然端粒延长。相比之下,X 连锁先天性角化不良症中 DKC1 (也称为 WRAP53)突变严重损害端粒酶活性,通过阻断端粒酶组装并破坏重编程过程中的端粒延长来破坏端粒酶活性。在由 TCAB1 (也称为 WRAP53)突变引起的先天性角化不良症的一种形式的 iPSC 中,端粒酶催化活性不受干扰,但端粒酶延长端粒的能力被阻断,因为端粒酶从 Cajal 体错误定位到 iPSC 中的核仁。DKC1 突变的 iPSC 的延长培养导致端粒逐渐缩短并最终丧失自我更新能力,表明在先天性角化不良症患者的组织干细胞中也发生了类似的过程。这些来自先天性角化不良症患者的 iPSC 的发现表明,未分化的 iPSC 准确地再现了人类干细胞疾病的特征,并且可以作为一种基于细胞培养的系统来开发靶向治疗方法。