Center for Bioinformatics, University of Hamburg, Bundesstrasse 43, 20146 Hamburg, Germany.
BMC Bioinformatics. 2011 May 27;12:214. doi: 10.1186/1471-2105-12-214.
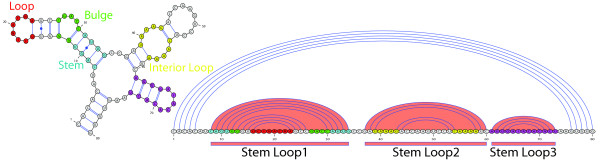
The secondary structure of RNA molecules is intimately related to their function and often more conserved than the sequence. Hence, the important task of searching databases for RNAs requires to match sequence-structure patterns. Unfortunately, current tools for this task have, in the best case, a running time that is only linear in the size of sequence databases. Furthermore, established index data structures for fast sequence matching, like suffix trees or arrays, cannot benefit from the complementarity constraints introduced by the secondary structure of RNAs.
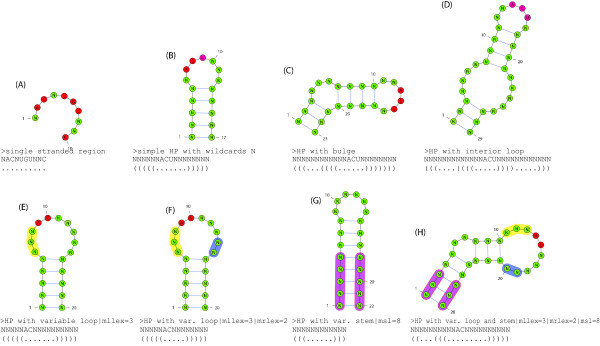
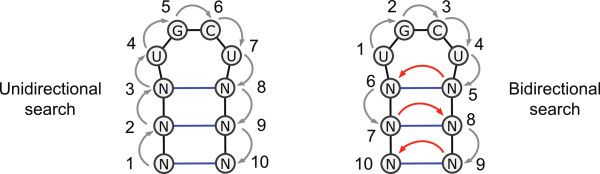
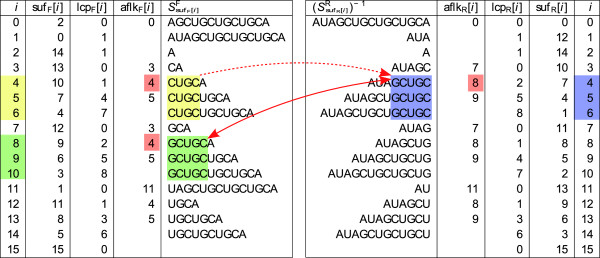
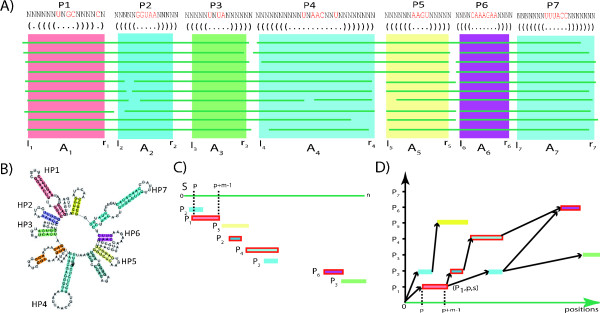
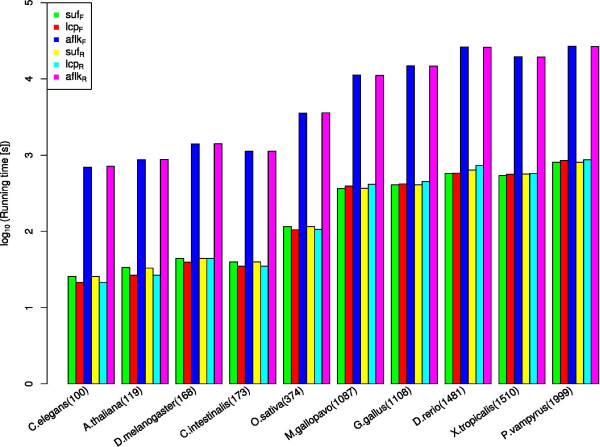
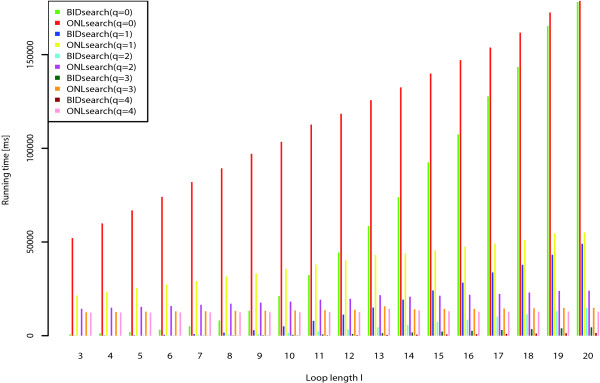
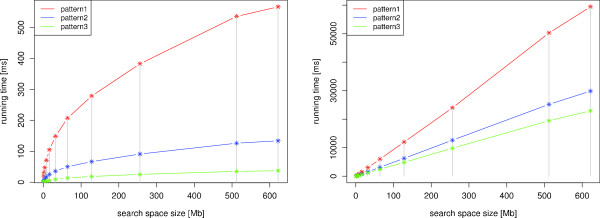
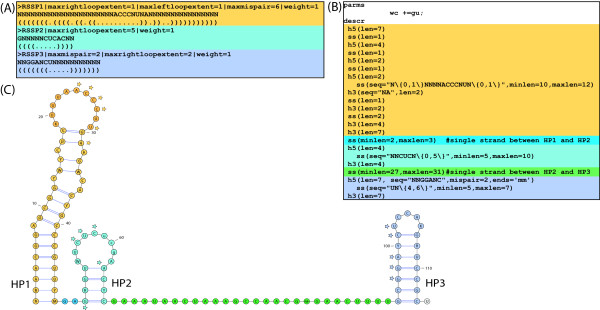
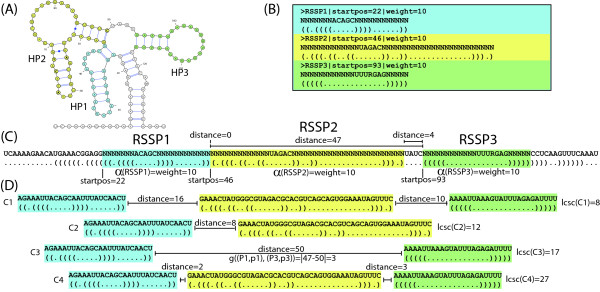
We present a novel method and readily applicable software for time efficient matching of RNA sequence-structure patterns in sequence databases. Our approach is based on affix arrays, a recently introduced index data structure, preprocessed from the target database. Affix arrays support bidirectional pattern search, which is required for efficiently handling the structural constraints of the pattern. Structural patterns like stem-loops can be matched inside out, such that the loop region is matched first and then the pairing bases on the boundaries are matched consecutively. This allows to exploit base pairing information for search space reduction and leads to an expected running time that is sublinear in the size of the sequence database. The incorporation of a new chaining approach in the search of RNA sequence-structure patterns enables the description of molecules folding into complex secondary structures with multiple ordered patterns. The chaining approach removes spurious matches from the set of intermediate results, in particular of patterns with little specificity. In benchmark experiments on the Rfam database, our method runs up to two orders of magnitude faster than previous methods.
The presented method's sublinear expected running time makes it well suited for RNA sequence-structure pattern matching in large sequence databases. RNA molecules containing several stem-loop substructures can be described by multiple sequence-structure patterns and their matches are efficiently handled by a novel chaining method. Beyond our algorithmic contributions, we provide with Structator a complete and robust open-source software solution for index-based search of RNA sequence-structure patterns. The Structator software is available at http://www.zbh.uni-hamburg.de/Structator.
RNA 分子的二级结构与其功能密切相关,通常比序列更保守。因此,在数据库中搜索 RNA 时,需要匹配序列-结构模式。不幸的是,目前用于此任务的工具在最佳情况下,其运行时间仅与序列数据库的大小呈线性关系。此外,用于快速序列匹配的现有索引数据结构(如后缀树或数组)不能受益于 RNA 二级结构引入的互补约束。
我们提出了一种新颖的方法和易于应用的软件,用于在序列数据库中高效匹配 RNA 序列-结构模式。我们的方法基于后缀数组,后缀数组是一种最近提出的索引数据结构,从目标数据库中预处理得到。后缀数组支持双向模式搜索,这是有效处理模式结构约束所必需的。像茎环这样的结构模式可以内外匹配,即首先匹配环区,然后连续匹配边界上的配对碱基。这允许利用碱基配对信息来缩小搜索空间,并导致运行时间与序列数据库的大小呈次线性关系。在搜索 RNA 序列-结构模式时采用新的链接方法,使得能够描述具有多个有序模式的复杂二级结构的分子折叠。链接方法从中间结果集中消除了虚假匹配,特别是那些特异性小的模式。在 Rfam 数据库上的基准实验中,我们的方法比以前的方法快两个数量级。
所提出的方法的预期运行时间呈次线性,非常适合在大型序列数据库中进行 RNA 序列-结构模式匹配。包含多个茎环亚结构的 RNA 分子可以通过多个序列-结构模式来描述,并且它们的匹配可以通过新的链接方法有效地处理。除了我们的算法贡献之外,我们还提供了 Structator,这是一个完整的、健壮的开源软件解决方案,用于基于索引的 RNA 序列-结构模式搜索。Structator 软件可在 http://www.zbh.uni-hamburg.de/Structator 上获得。