Department of Biology, Bologna Biocomputing Group, Bologna Computational Biology Network, Bologna, Italy.
Nucleic Acids Res. 2011 Jul;39(Web Server issue):W197-202. doi: 10.1093/nar/gkr292. Epub 2011 May 26.
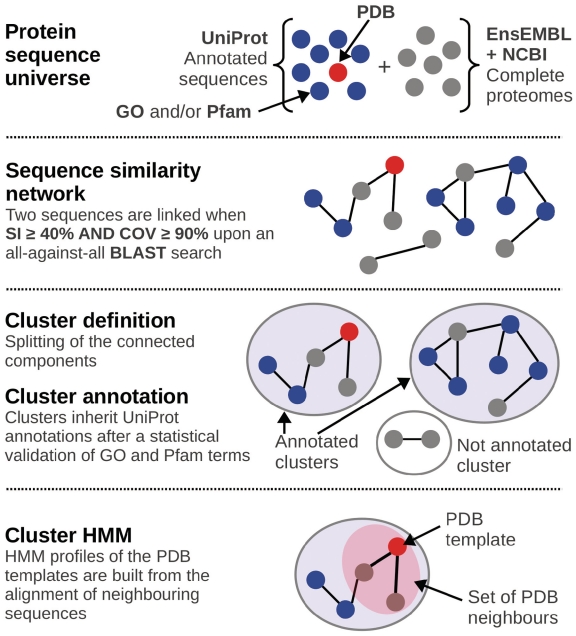
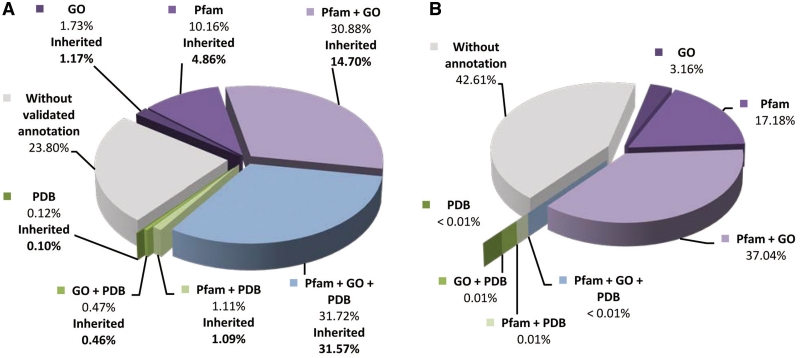
We introduce BAR-PLUS (BAR(+)), a web server for functional and structural annotation of protein sequences. BAR(+) is based on a large-scale genome cross comparison and a non-hierarchical clustering procedure characterized by a metric that ensures a reliable transfer of features within clusters. In this version, the method takes advantage of a large-scale pairwise sequence comparison of 13,495,736 protein chains also including 988 complete proteomes. Available sequence annotation is derived from UniProtKB, GO, Pfam and PDB. When PDB templates are present within a cluster (with or without their SCOP classification), profile Hidden Markov Models (HMMs) are computed on the basis of sequence to structure alignment and are cluster-associated (Cluster-HMM). Therefrom, a library of 10,858 HMMs is made available for aligning even distantly related sequences for structural modelling. The server also provides pairwise query sequence-structural target alignments computed from the correspondent Cluster-HMM. BAR(+) in its present version allows three main categories of annotation: PDB [with or without SCOP ()] and GO and/or Pfam; PDB () without GO and/or Pfam; GO and/or Pfam without PDB (*) and no annotation. Each category can further comprise clusters where GO and Pfam functional annotations are or are not statistically significant. BAR(+) is available at http://bar.biocomp.unibo.it/bar2.0.
我们介绍了 BAR-PLUS(BAR(+)),这是一个用于蛋白质序列功能和结构注释的网络服务器。BAR(+) 基于大规模的基因组交叉比较和非层次聚类过程,该过程的特点是采用一种度量标准,确保在聚类内部可靠地传递特征。在这个版本中,该方法利用了对 13495736 条蛋白质链的大规模两两序列比较,其中还包括 988 个完整的蛋白质组。可用的序列注释来自 UniProtKB、GO、Pfam 和 PDB。当 PDB 模板存在于一个聚类中(无论是否具有 SCOP 分类)时,会基于序列到结构的比对计算轮廓隐马尔可夫模型(HMM),并与聚类相关联(Cluster-HMM)。由此,提供了一个包含 10858 个 HMM 的库,用于对齐甚至远缘相关的序列进行结构建模。该服务器还提供了基于对应 Cluster-HMM 的两两查询序列-结构目标比对。BAR(+) 在其当前版本中允许三种主要的注释类别:PDB [带有或不带有 SCOP() ] 和 GO 和/或 Pfam;PDB() 不带 GO 和/或 Pfam;GO 和/或 Pfam 不带 PDB(*) 且没有注释。每个类别还可以进一步包含 GO 和 Pfam 功能注释是否具有统计学意义的聚类。BAR(+) 可在 http://bar.biocomp.unibo.it/bar2.0 访问。