Marine Biology Research Division, Scripps Institution of Oceanography, University of California San Diego, La Jolla, California, United States of America.
PLoS One. 2011;6(5):e20388. doi: 10.1371/journal.pone.0020388. Epub 2011 May 24.
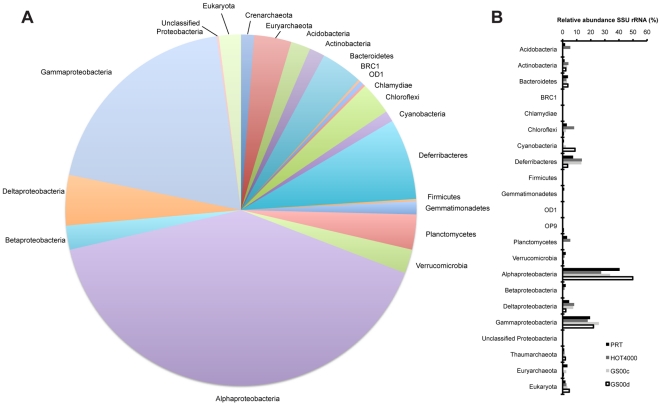
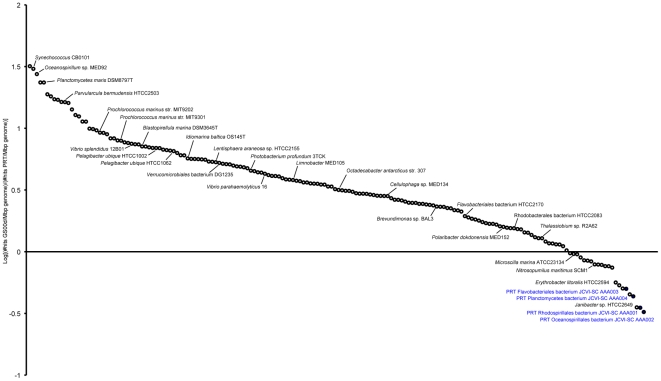
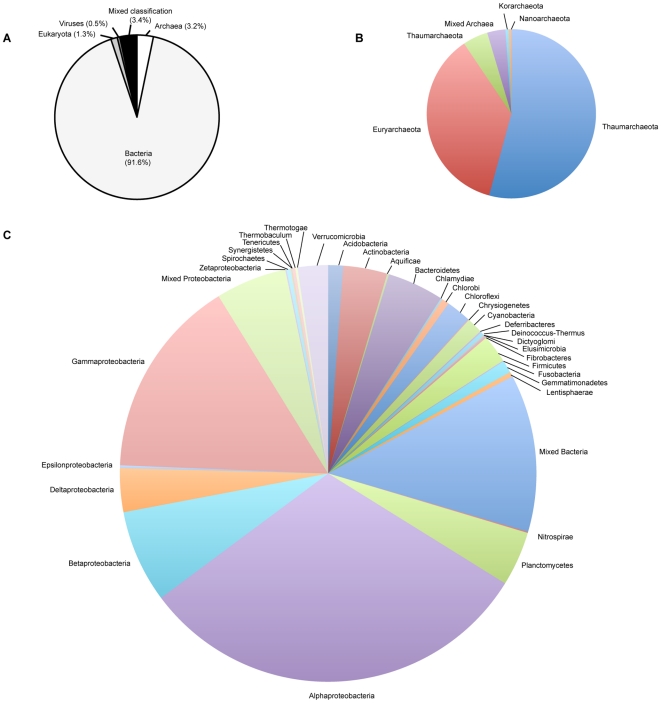
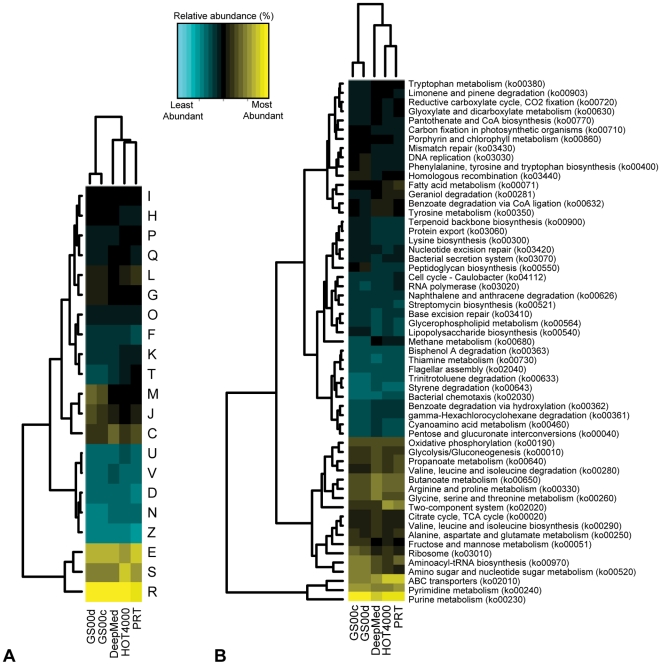
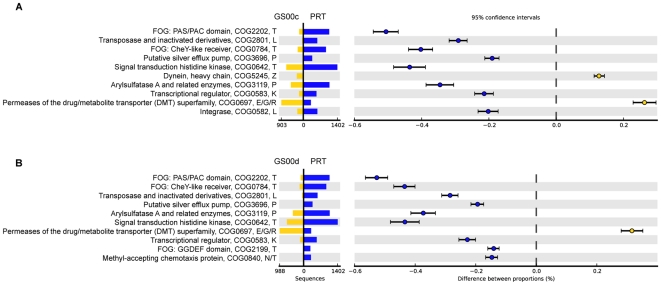
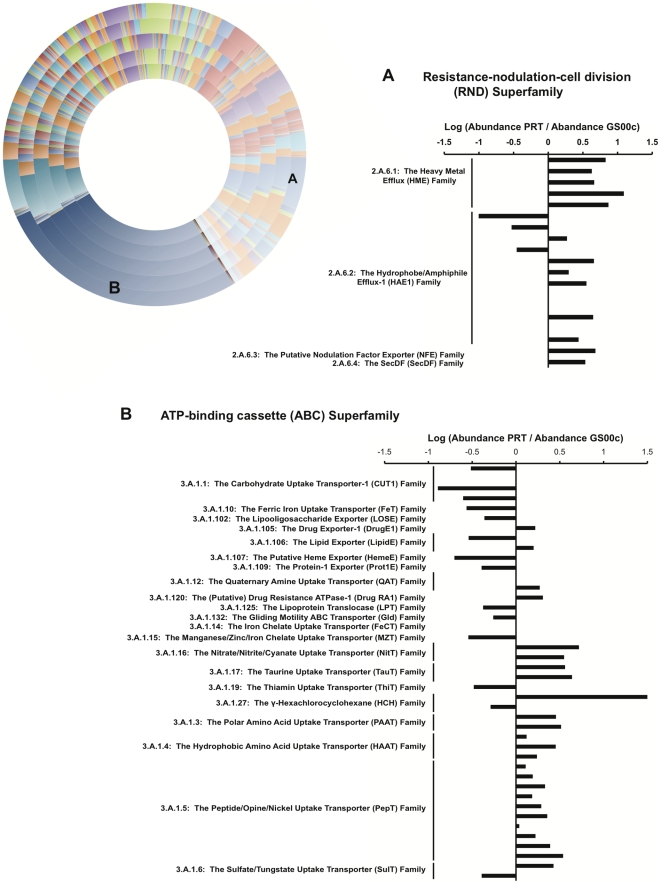
The paucity of sequence data from pelagic deep-ocean microbial assemblages has severely restricted molecular exploration of the largest biome on Earth. In this study, an analysis is presented of a large-scale 454-pyrosequencing metagenomic dataset from a hadopelagic environment from 6,000 m depth within the Puerto Rico Trench (PRT). A total of 145 Mbp of assembled sequence data was generated and compared to two pelagic deep ocean metagenomes and two representative surface seawater datasets from the Sargasso Sea. In a number of instances, all three deep metagenomes displayed similar trends, but were most magnified in the PRT, including enrichment in functions for two-component signal transduction mechanisms and transcriptional regulation. Overrepresented transporters in the PRT metagenome included outer membrane porins, diverse cation transporters, and di- and tri-carboxylate transporters that matched well with the prevailing catabolic processes such as butanoate, glyoxylate and dicarboxylate metabolism. A surprisingly high abundance of sulfatases for the degradation of sulfated polysaccharides were also present in the PRT. The most dramatic adaptational feature of the PRT microbes appears to be heavy metal resistance, as reflected in the large numbers of transporters present for their removal. As a complement to the metagenome approach, single-cell genomic techniques were utilized to generate partial whole-genome sequence data from four uncultivated cells from members of the dominant phyla within the PRT, Alphaproteobacteria, Gammaproteobacteria, Bacteroidetes and Planctomycetes. The single-cell sequence data provided genomic context for many of the highly abundant functional attributes identified from the PRT metagenome, as well as recruiting heavily the PRT metagenomic sequence data compared to 172 available reference marine genomes. Through these multifaceted sequence approaches, new insights have been provided into the unique functional attributes present in microbes residing in a deeper layer of the ocean far removed from the more productive sun-drenched zones above.
海洋深处微生物组合的序列数据的匮乏,严重限制了对地球上最大生物群落的分子探索。在这项研究中,对来自波多黎各海沟(PRT) 6000 米深处的海洋上层环境的大规模 454 焦磷酸测序宏基因组数据集进行了分析。总共生成了 145 Mbp 的组装序列数据,并与两个海洋深层宏基因组和两个来自马尾藻海的代表性表层海水数据集进行了比较。在许多情况下,所有三个深海宏基因组都显示出相似的趋势,但在 PRT 中最为明显,包括对双组分信号转导机制和转录调控的功能富集。PRT 宏基因组中过度表达的转运蛋白包括外膜孔蛋白、多种阳离子转运蛋白和二羧酸和三羧酸转运蛋白,与丁酸盐、乙醛酸盐和二羧酸代谢等流行的分解代谢过程非常匹配。PRT 中还存在大量用于降解硫酸多糖的硫酸酯酶。PRT 微生物的一个惊人的适应性特征似乎是重金属抗性,这反映在存在大量用于去除重金属的转运蛋白。作为宏基因组方法的补充,单细胞基因组技术被用于从 PRT 中占优势的门的四个未培养细胞中生成部分全基因组序列数据,这些门包括α变形菌门、γ变形菌门、拟杆菌门和浮霉菌门。单细胞序列数据为 PRT 宏基因组中许多高度丰富的功能属性提供了基因组背景,并且与 172 个可用的参考海洋基因组相比,大量招募了 PRT 宏基因组序列数据。通过这些多方面的序列方法,我们对生活在远离阳光充足的上层更具生产力的海洋深层的微生物的独特功能属性有了新的认识。