Program in Developmental Biology, Baylor College of Medicine, Houston, TX 77030, USA.
Science. 2011 Jul 8;333(6039):186. doi: 10.1126/science.1206593. Epub 2011 Jun 2.
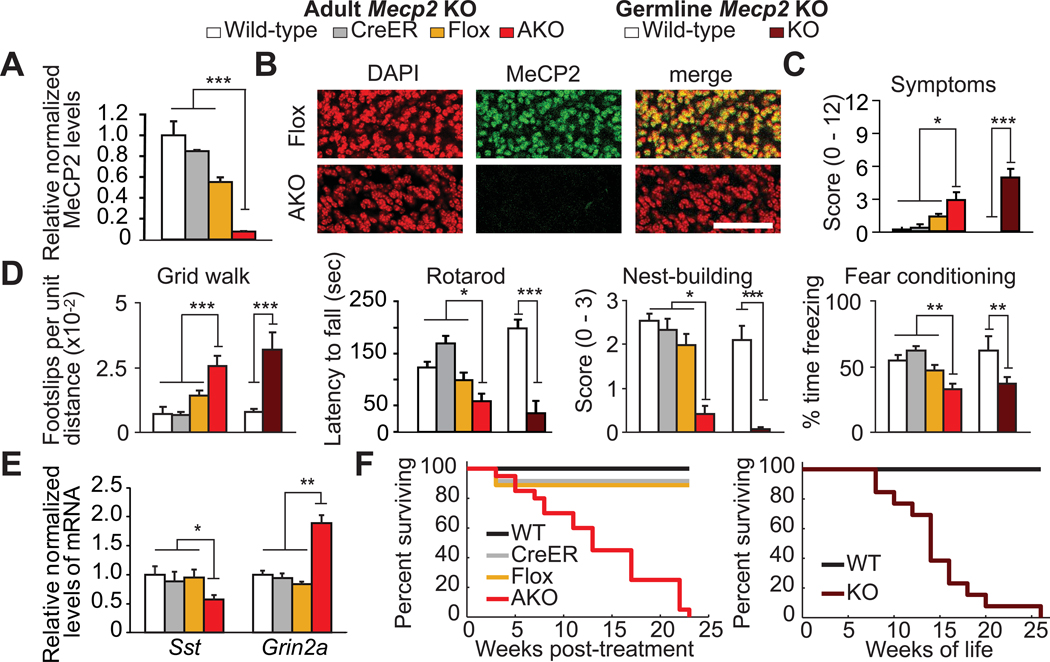
Rett syndrome (RTT) is a postnatal neurological disorder caused by mutations in MECP2, encoding the epigenetic regulator methyl-CpG-binding protein 2 (MeCP2). The onset of RTT symptoms during early life together with findings suggesting neurodevelopmental abnormalities in RTT and mouse models of RTT raised the question of whether maintaining MeCP2 function exclusively during early life might protect against disease. We show by using an inducible model of RTT that deletion of Mecp2 in adult mice recapitulates the germline knock-out phenotype, underscoring the ongoing role of MeCP2 in adult neurological function. Moreover, unlike the effects of other epigenetic instructions programmed during early life, the effects of early MeCP2 function are lost soon after its deletion. These findings suggest that therapies for RTT must be maintained throughout life.
雷特综合征(RTT)是一种后天性神经发育障碍,由 MECP2 基因突变引起,该基因编码表观遗传调控因子甲基化CpG 结合蛋白 2(MeCP2)。RTT 症状在生命早期出现,同时有研究表明 RTT 患者和 RTT 小鼠模型存在神经发育异常,这引发了一个问题,即在生命早期仅维持 MeCP2 功能是否可以预防疾病。我们通过使用 RTT 的诱导模型表明,成年小鼠中 Mecp2 的缺失重现了种系敲除表型,突出了 MeCP2 在成年神经功能中的持续作用。此外,与生命早期编程的其他表观遗传指令的影响不同,早期 MeCP2 功能的影响在其缺失后很快就会消失。这些发现表明,RTT 的治疗必须终身维持。