Department of Genome Sciences, University of Washington, Seattle, Washington 98195, USA.
Proteins. 2011 Aug;79(8):2380-8. doi: 10.1002/prot.23046. Epub 2011 Jun 2.
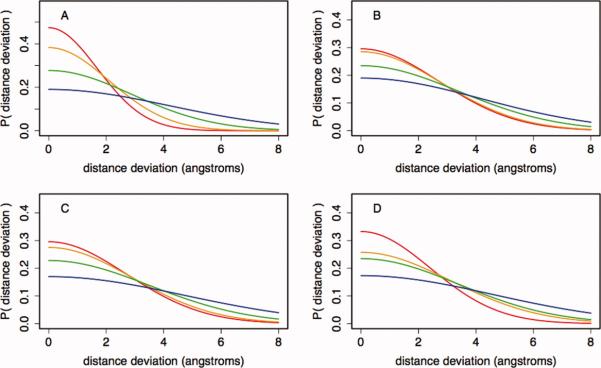
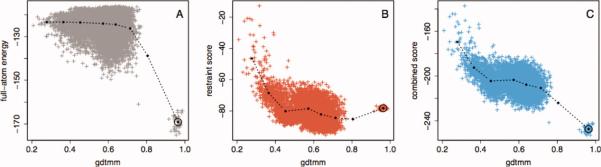
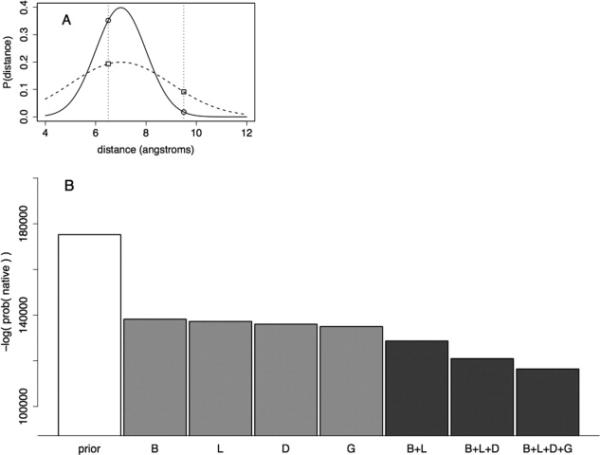
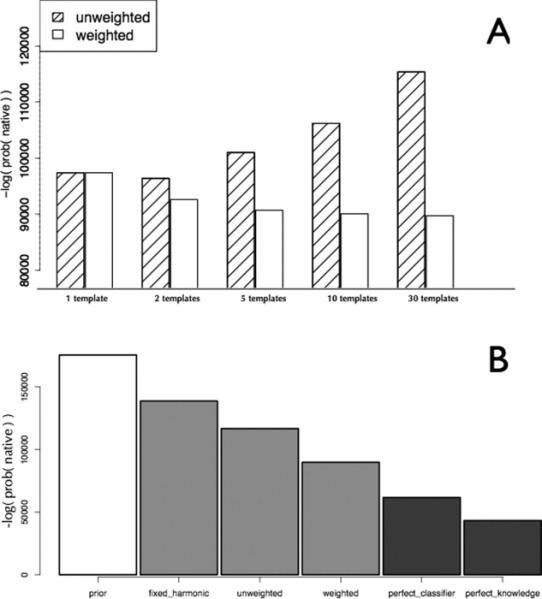
Prediction of protein structures from sequences is a fundamental problem in computational biology. Algorithms that attempt to predict a structure from sequence primarily use two sources of information. The first source is physical in nature: proteins fold into their lowest energy state. Given an energy function that describes the interactions governing folding, a method for constructing models of protein structures, and the amino acid sequence of a protein of interest, the structure prediction problem becomes a search for the lowest energy structure. Evolution provides an orthogonal source of information: proteins of similar sequences have similar structure, and therefore proteins of known structure can guide modeling. The relatively successful Rosetta approach takes advantage of the first, but not the second source of information during model optimization. Following the classic work by Andrej Sali and colleagues, we develop a probabilistic approach to derive spatial restraints from proteins of known structure using advances in alignment technology and the growth in the number of structures in the Protein Data Bank. These restraints define a region of conformational space that is high-probability, given the template information, and we incorporate them into Rosetta's comparative modeling protocol. The combined approach performs considerably better on a benchmark based on previous CASP experiments. Incorporating evolutionary information into Rosetta is analogous to incorporating sparse experimental data: in both cases, the additional information eliminates large regions of conformational space and increases the probability that energy-based refinement will hone in on the deep energy minimum at the native state.
从序列预测蛋白质结构是计算生物学中的一个基本问题。尝试从序列预测结构的算法主要使用两种信息来源。第一个来源是物理性质:蛋白质折叠成其最低能量状态。给定一个描述折叠控制相互作用的能量函数、构建蛋白质结构模型的方法以及感兴趣的蛋白质的氨基酸序列,结构预测问题就变成了寻找最低能量结构的问题。进化提供了另一个信息来源:具有相似序列的蛋白质具有相似的结构,因此具有已知结构的蛋白质可以指导建模。相对成功的 Rosetta 方法在模型优化过程中利用了第一个来源,但没有利用第二个来源的信息。受 Andrej Sali 及其同事的经典工作的启发,我们利用对齐技术的进步和蛋白质数据库中结构数量的增加,从已知结构的蛋白质中推导出空间限制,从而开发出一种从概率角度推导空间限制的方法。这些限制定义了一个构象空间区域,在给定模板信息的情况下,该区域具有高概率,我们将其纳入 Rosetta 的比较建模协议中。这种组合方法在基于以前 CASP 实验的基准测试中表现要好得多。将进化信息纳入 Rosetta 类似于将稀疏实验数据纳入其中:在这两种情况下,额外的信息都会消除构象空间的大部分区域,并增加基于能量的细化将聚焦于天然状态下的深能量最低点的概率。