Division of Maternal-Fetal Medicine and Laboratory of Perinatal Research, Department of Obstetrics and Gynecology, The Ohio State University, Columbus, Ohio, United States of America.
PLoS One. 2011;6(6):e20560. doi: 10.1371/journal.pone.0020560. Epub 2011 Jun 2.
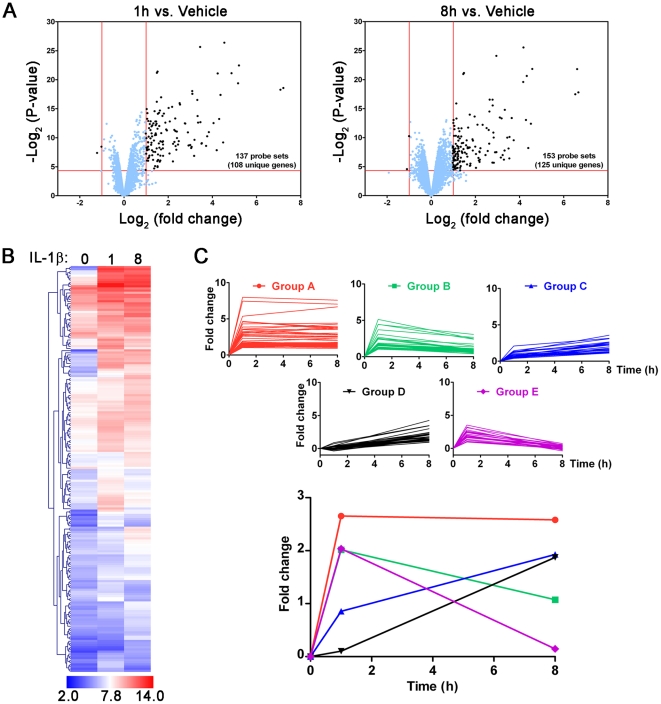
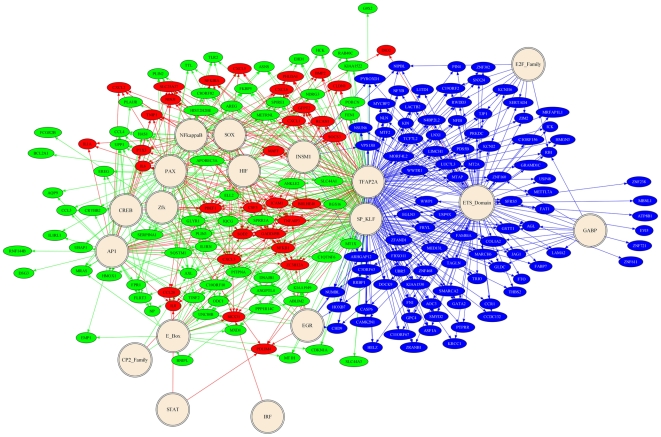
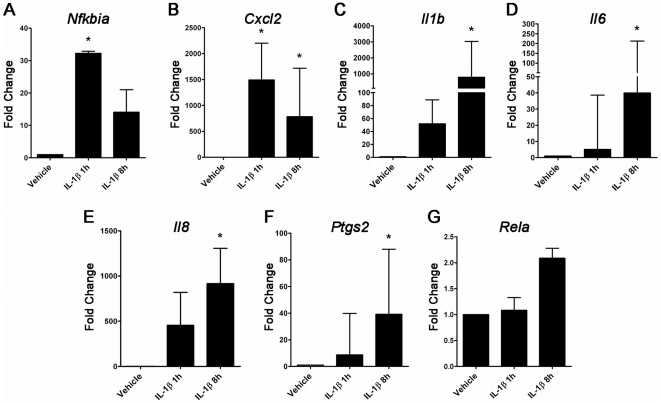
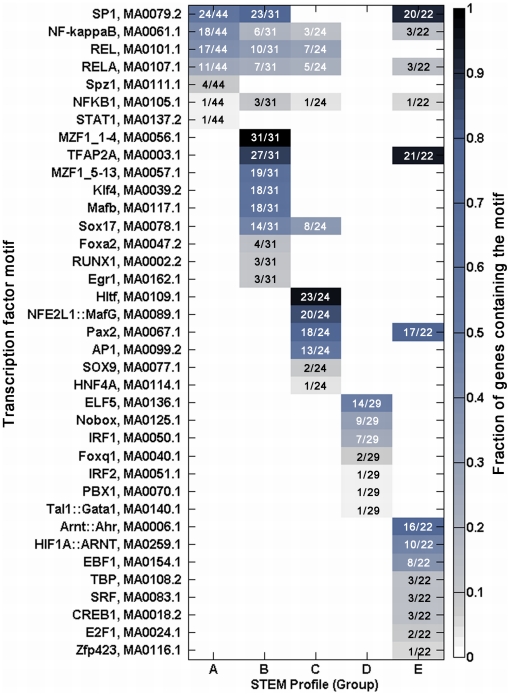
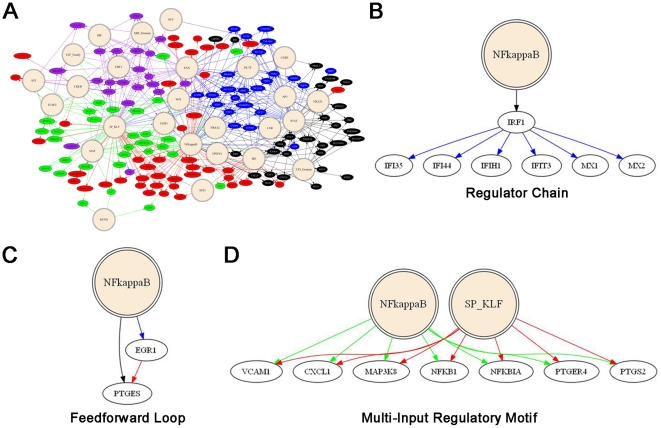
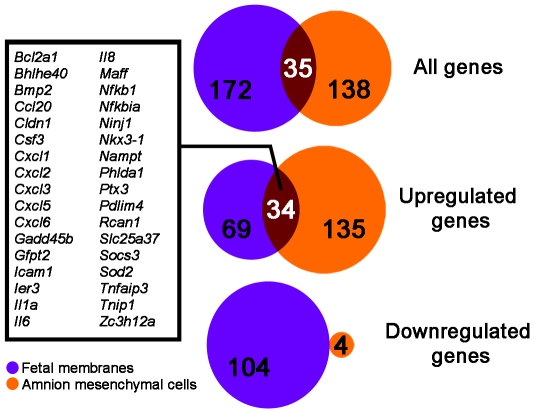
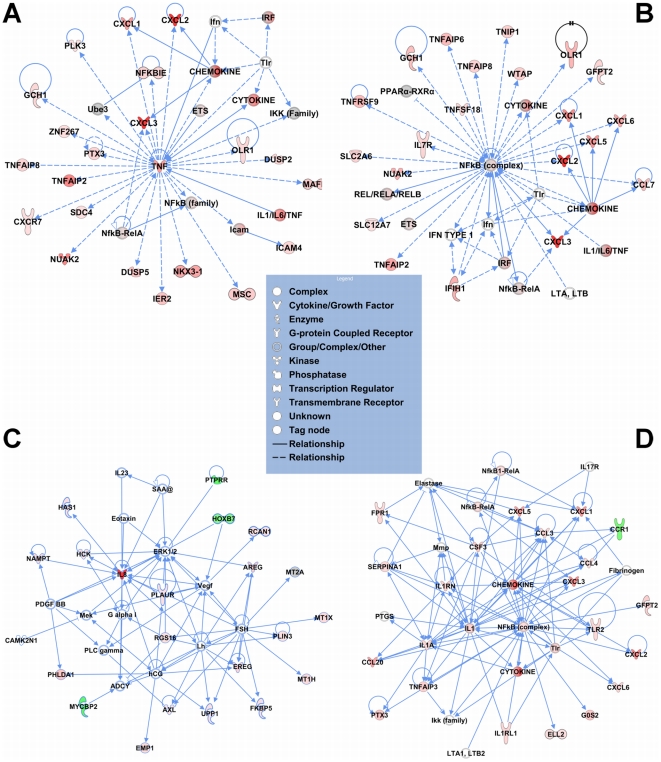
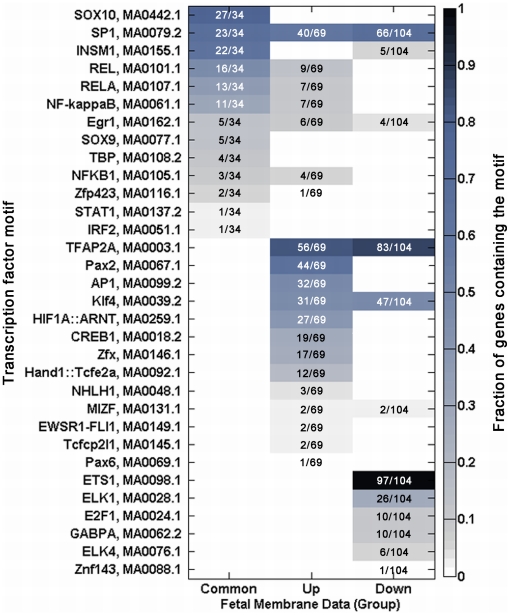
A majority of the studies examining the molecular regulation of human labor have been conducted using single gene approaches. While the technology to produce multi-dimensional datasets is readily available, the means for facile analysis of such data are limited. The objective of this study was to develop a systems approach to infer regulatory mechanisms governing global gene expression in cytokine-challenged cells in vitro, and to apply these methods to predict gene regulatory networks (GRNs) in intrauterine tissues during term parturition. To this end, microarray analysis was applied to human amnion mesenchymal cells (AMCs) stimulated with interleukin-1β, and differentially expressed transcripts were subjected to hierarchical clustering, temporal expression profiling, and motif enrichment analysis, from which a GRN was constructed. These methods were then applied to fetal membrane specimens collected in the absence or presence of spontaneous term labor. Analysis of cytokine-responsive genes in AMCs revealed a sterile immune response signature, with promoters enriched in response elements for several inflammation-associated transcription factors. In comparison to the fetal membrane dataset, there were 34 genes commonly upregulated, many of which were part of an acute inflammation gene expression signature. Binding motifs for nuclear factor-κB were prominent in the gene interaction and regulatory networks for both datasets; however, we found little evidence to support the utilization of pathogen-associated molecular pattern (PAMP) signaling. The tissue specimens were also enriched for transcripts governed by hypoxia-inducible factor. The approach presented here provides an uncomplicated means to infer global relationships among gene clusters involved in cellular responses to labor-associated signals.
大多数研究人类分娩的分子调控机制的研究都是采用单一基因方法进行的。虽然生成多维数据集的技术已经很成熟,但分析这些数据的方法却很有限。本研究的目的是开发一种系统方法,以推断体外细胞因子刺激下细胞中控制全局基因表达的调控机制,并将这些方法应用于预测足月分娩时宫内组织中的基因调控网络(GRN)。为此,对白细胞介素 1β刺激的人羊膜间充质细胞(AMCs)进行了微阵列分析,并对差异表达的转录本进行了层次聚类、时间表达谱分析和基序富集分析,从而构建了一个 GRN。然后将这些方法应用于收集的无自发性足月分娩和有自发性足月分娩的胎膜标本。对 AMCs 中细胞因子反应基因的分析显示出一种无菌免疫反应特征,其启动子富含几种与炎症相关的转录因子的反应元件。与胎膜数据集相比,有 34 个基因共同上调,其中许多基因是急性炎症基因表达特征的一部分。核因子-κB 的结合基序在两个数据集的基因相互作用和调控网络中都很突出;然而,我们几乎没有证据支持病原体相关分子模式(PAMP)信号的利用。组织标本中还富含受缺氧诱导因子调控的转录本。这里提出的方法提供了一种简单的方法来推断与分娩相关信号的细胞反应中涉及的基因簇之间的全局关系。