Gladstone Institutes, University of California, San Francisco, USA.
Genome Biol Evol. 2011;3:516-27. doi: 10.1093/gbe/evr051. Epub 2011 Jun 13.
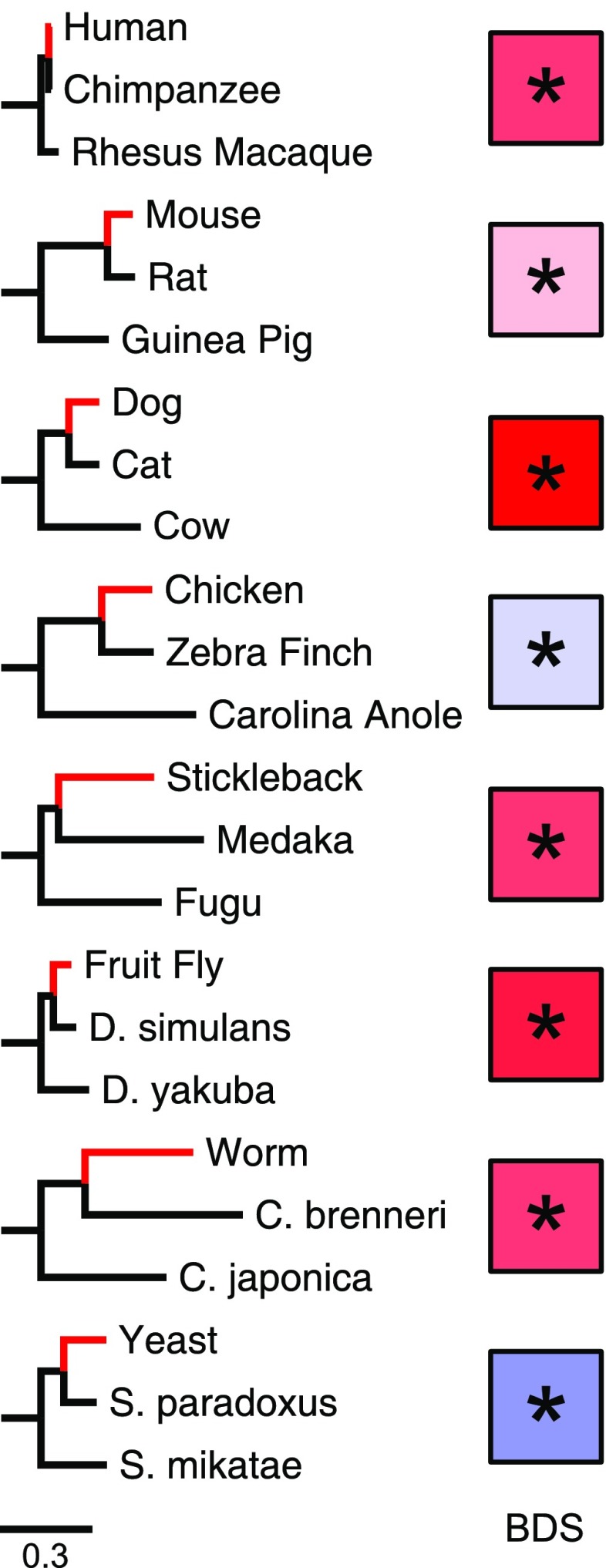
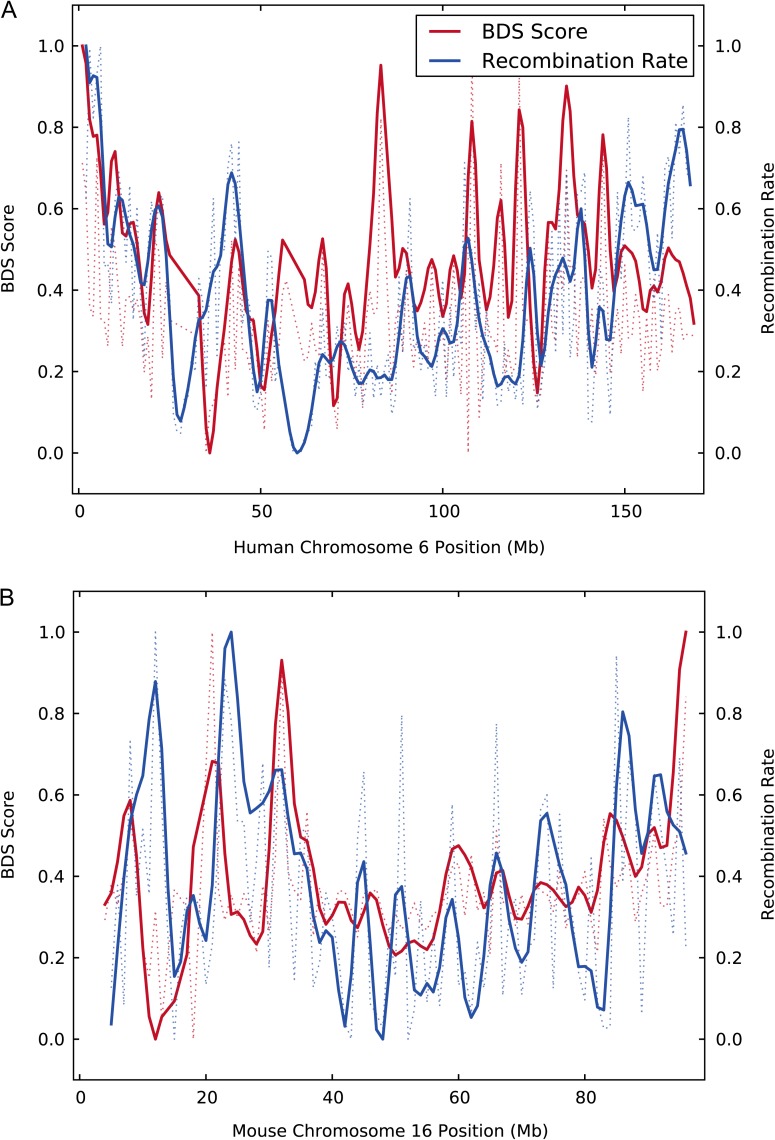
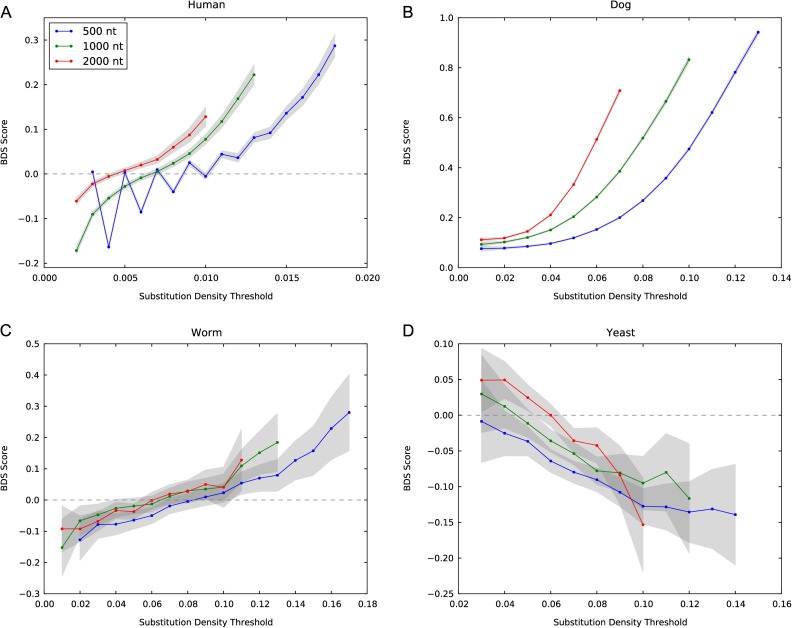
The fastest-evolving regions in the human and chimpanzee genomes show a remarkable excess of weak (A,T) to strong (G,C) nucleotide substitutions since divergence from their common ancestor. We investigated the phylogenetic extent and possible causes of this weak to strong (W → S) bias in divergent sequences (BDS) using recently sequenced genomes and recombination maps from eight trios of eukaryotic species. To quantify evidence for BDS, we inferred substitution histories using an efficient maximum likelihood approach with a context-dependent evolutionary model. We then annotated all lineage-specific substitutions in terms of W → S bias and density on the chromosomes. Finally, we used the inferred substitutions to calculate a BDS score-a log odds ratio between substitution type and density-and assessed its statistical significance with Fisher's exact test. Applying this approach, we found significant BDS in the coding and noncoding sequence of human, mouse, dog, stickleback, fruit fly, and worm. We also observed a significant lack of W → S BDS in chicken and yeast. The BDS score varies between species and across the chromosomes within each species. It is most strongly correlated with different genomic features in different species, but a strong correlation with recombination rates is found in several species. Our results demonstrate that a W → S substitution bias in fast-evolving sequences is a widespread phenomenon. The patterns of BDS observed suggest that a recombination-associated process, such as GC-biased gene conversion, is involved in the production of the bias in many species, but the strength of the BDS likely depends on many factors, including genome stability, variability in recombination rate over time and across the genome, the frequency of meiosis, and the amount of outcrossing in each species.
在人类和黑猩猩基因组中进化最快的区域,自与共同祖先分化以来,显示出明显的弱(A,T)到强(G,C)核苷酸取代过度。我们使用最近测序的基因组和来自八个真核生物种系的重组图谱,研究了这种在分歧序列(BDS)中从弱到强(W→S)偏倚的系统发育范围和可能的原因。为了量化 BDS 的证据,我们使用具有上下文相关进化模型的高效最大似然方法推断取代历史。然后,我们根据 W→S 偏倚和染色体上的密度来注释所有谱系特异性取代。最后,我们使用推断出的取代来计算 BDS 分数 - 取代类型和密度之间的对数优势比 - 并使用 Fisher 精确检验评估其统计显著性。应用这种方法,我们在人类、小鼠、狗、棘鱼、果蝇和蠕虫的编码和非编码序列中发现了显著的 BDS。我们还观察到鸡和酵母中存在显著缺乏 W→S BDS。BDS 分数在物种之间以及每个物种的染色体之间变化。它与不同物种的不同基因组特征密切相关,但在几个物种中发现与重组率有很强的相关性。我们的研究结果表明,快速进化序列中的 W→S 取代偏倚是一种普遍现象。观察到的 BDS 模式表明,重组相关过程,如 GC 偏向性基因转换,参与了许多物种中偏倚的产生,但 BDS 的强度可能取决于许多因素,包括基因组稳定性、重组率随时间和整个基因组的变化、减数分裂的频率以及每个物种的杂交程度。