Department of Electrical and Computer Engineering, University of Alberta, Edmonton, Canada.
Bioinformatics. 2011 Jul 1;27(13):i24-33. doi: 10.1093/bioinformatics/btr229.
X-ray crystallography-based protein structure determination, which accounts for majority of solved structures, is characterized by relatively low success rates. One solution is to build tools which support selection of targets that are more likely to crystallize. Several in silico methods that predict propensity of diffraction-quality crystallization from protein chains were developed. We show that the quality of their predictions drops when applied to more recent crystallization trails, which calls for new solutions. We propose a novel approach that alleviates drawbacks of the existing methods by using a recent dataset and improved protocol to annotate progress along the crystallization process, by predicting the success of the entire process and steps which result in the failed attempts, and by utilizing a compact and comprehensive set of sequence-derived inputs to generate accurate predictions.
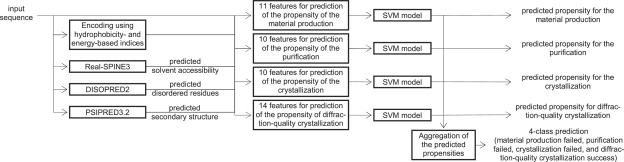
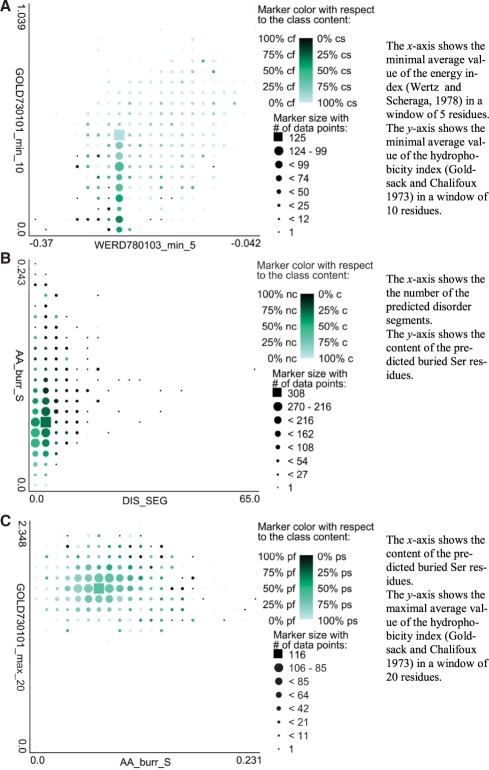
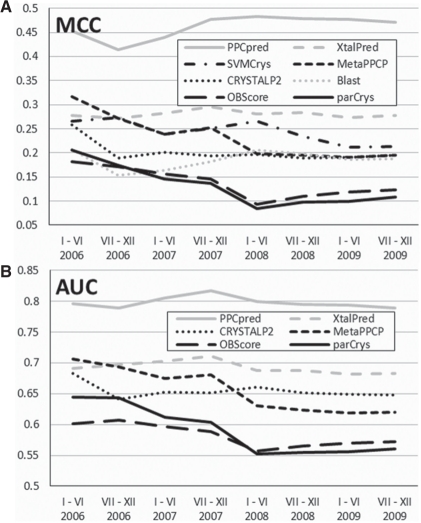
The proposed PPCpred (predictor of protein Production, Purification and Crystallization) predict propensity for production of diffraction-quality crystals, production of crystals, purification and production of the protein material. PPCpred utilizes comprehensive set of inputs based on energy and hydrophobicity indices, composition of certain amino acid types, predicted disorder, secondary structure and solvent accessibility, and content of certain buried and exposed residues. Our method significantly outperforms alignment-based predictions and several modern crystallization propensity predictors. Receiver operating characteristic (ROC) curves show that PPCpred is particularly useful for users who desire high true positive (TP) rates, i.e. low rate of mispredictions for solvable chains. Our model reveals several intuitive factors that influence the success of individual steps and the entire crystallization process, including the content of Cys, buried His and Ser, hydrophobic/hydrophilic segments and the number of predicted disordered segments.
http://biomine.ece.ualberta.ca/PPCpred/.
基于 X 射线晶体学的蛋白质结构测定,占已解决结构的大多数,其成功率相对较低。一种解决方案是构建支持选择更有可能结晶的目标的工具。已经开发了几种从蛋白质链预测衍射质量结晶倾向的计算方法。我们表明,当将它们应用于最近的结晶试验时,它们的预测质量会下降,这需要新的解决方案。我们提出了一种新方法,通过使用最近的数据集和改进的方案来注释结晶过程的进展,通过预测整个过程的成功以及导致失败尝试的步骤,并利用紧凑而全面的基于序列的输入集来生成准确的预测,从而减轻现有方法的缺点。
所提出的 PPCpred(蛋白质生产、纯化和结晶预测器)预测了生产衍射质量晶体、生产晶体、纯化和生产蛋白质材料的倾向。PPCpred 利用了基于能量和疏水性指数、某些氨基酸类型的组成、预测的无序、二级结构和溶剂可及性以及某些埋藏和暴露残基的含量的综合输入集。我们的方法显著优于基于比对的预测和几种现代结晶倾向预测器。接收者操作特征 (ROC) 曲线表明,PPCpred 对于希望获得高真阳性 (TP) 率的用户特别有用,即对于可解决链的错误预测率低。我们的模型揭示了几个直观的因素,这些因素会影响单个步骤和整个结晶过程的成功,包括 Cys、埋藏 His 和 Ser、疏水区/亲水区段以及预测的无序区段的数量。
http://biomine.ece.ualberta.ca/PPCpred/。