Program in Molecular and Computational Biology, University of Southern California, Los Angeles, CA, USA.
Bioinformatics. 2011 Jul 1;27(13):i401-9. doi: 10.1093/bioinformatics/btr206.
It is well known that microRNAs (miRNAs) and genes work cooperatively to form the key part of gene regulatory networks. However, the specific functional roles of most miRNAs and their combinatorial effects in cellular processes are still unclear. The availability of multiple types of functional genomic data provides unprecedented opportunities to study the miRNA-gene regulation. A major challenge is how to integrate the diverse genomic data to identify the regulatory modules of miRNAs and genes.
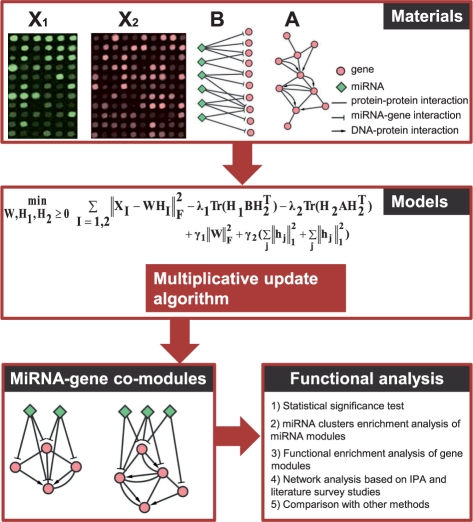
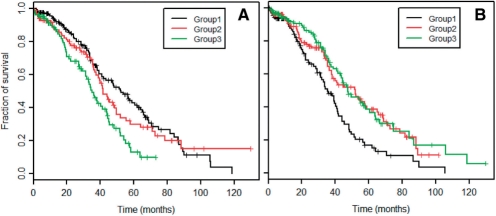
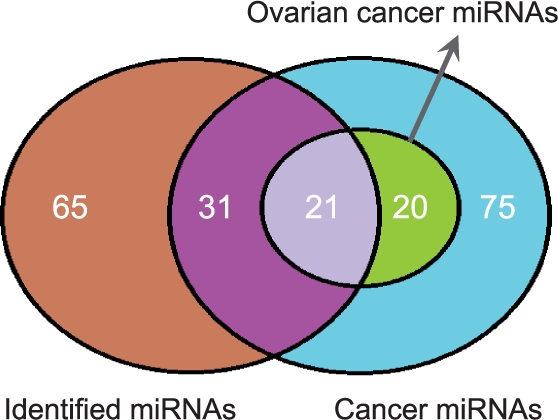
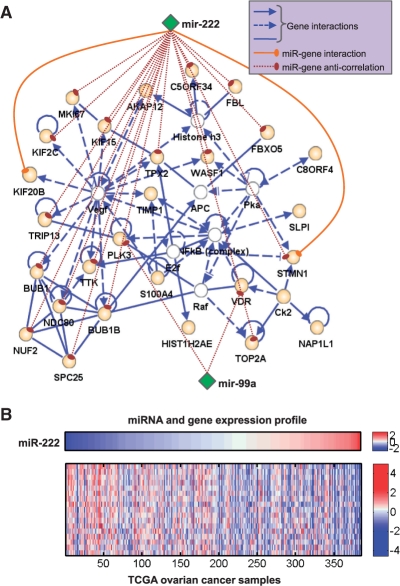
Here we propose an effective data integration framework to identify the miRNA-gene regulatory comodules. The miRNA and gene expression profiles are jointly analyzed in a multiple non-negative matrix factorization framework, and additional network data are simultaneously integrated in a regularized manner. Meanwhile, we employ the sparsity penalties to the variables to achieve modular solutions. The mathematical formulation can be effectively solved by an iterative multiplicative updating algorithm. We apply the proposed method to integrate a set of heterogeneous data sources including the expression profiles of miRNAs and genes on 385 human ovarian cancer samples, computationally predicted miRNA-gene interactions, and gene-gene interactions. We demonstrate that the miRNAs and genes in 69% of the regulatory comodules are significantly associated. Moreover, the comodules are significantly enriched in known functional sets such as miRNA clusters, GO biological processes and KEGG pathways, respectively. Furthermore, many miRNAs and genes in the comodules are related with various cancers including ovarian cancer. Finally, we show that comodules can stratify patients (samples) into groups with significant clinical characteristics.
The program and supplementary materials are available at http://zhoulab.usc.edu/SNMNMF/.
众所周知,microRNAs(miRNAs)和基因协同工作,形成基因调控网络的关键部分。然而,大多数 miRNAs 的具体功能作用及其在细胞过程中的组合效应仍不清楚。多种类型的功能基因组数据的可用性为研究 miRNA-基因调控提供了前所未有的机会。主要挑战是如何整合多样化的基因组数据,以识别 miRNAs 和基因的调控模块。
在这里,我们提出了一种有效的数据集成框架,用于识别 miRNA-基因的调控共模块。在多个非负矩阵分解框架中联合分析 miRNA 和基因表达谱,并以正则化的方式同时整合其他网络数据。同时,我们对变量使用稀疏惩罚以实现模块化解决方案。该数学公式可以通过迭代乘法更新算法有效地解决。我们应用所提出的方法来整合一组包括 385 个人类卵巢癌样本的 miRNA 和基因表达谱、计算预测的 miRNA-基因相互作用以及基因-基因相互作用的异构数据源。我们证明,调控共模块中 69%的 miRNAs 和基因显著相关。此外,共模块分别在已知的功能集(如 miRNA 簇、GO 生物学过程和 KEGG 途径)中显著富集。此外,共模块中的许多 miRNAs 和基因与包括卵巢癌在内的各种癌症有关。最后,我们表明共模块可以将患者(样本)分为具有显著临床特征的组。
程序和补充材料可在 http://zhoulab.usc.edu/SNMNMF/ 获得。