Program in Molecular and Computational Biology, Department of Biological Sciences, University of Southern California, Los Angeles, CA 90089, USA.
Bioinformatics. 2012 Oct 1;28(19):2458-66. doi: 10.1093/bioinformatics/bts476. Epub 2012 Aug 3.
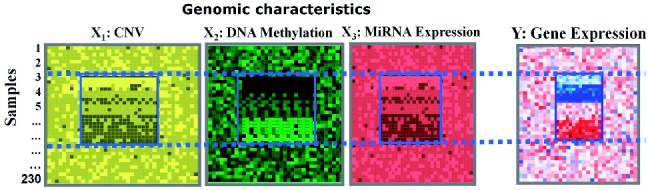
Eukaryotic gene expression (GE) is subjected to precisely coordinated multi-layer controls, across the levels of epigenetic, transcriptional and post-transcriptional regulations. Recently, the emerging multi-dimensional genomic dataset has provided unprecedented opportunities to study the cross-layer regulatory interplay. In these datasets, the same set of samples is profiled on several layers of genomic activities, e.g. copy number variation (CNV), DNA methylation (DM), GE and microRNA expression (ME). However, suitable analysis methods for such data are currently sparse.
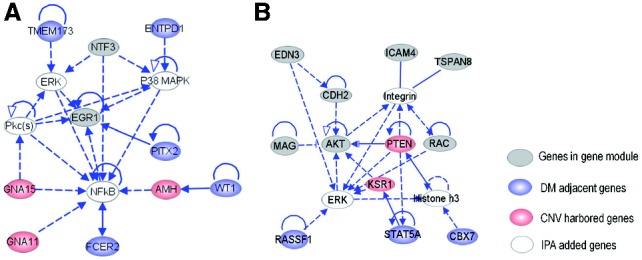
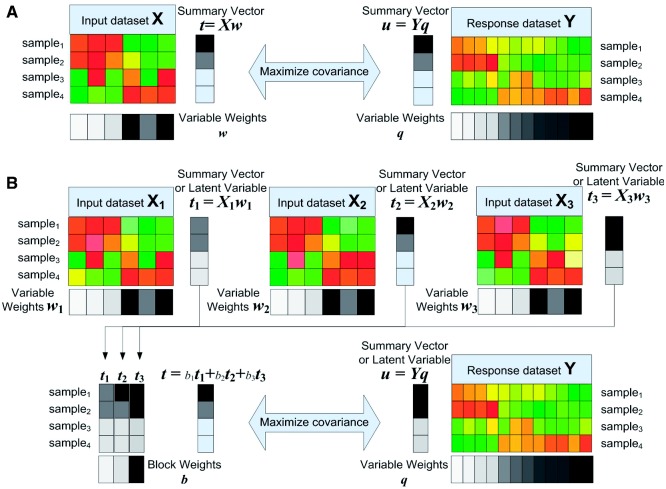
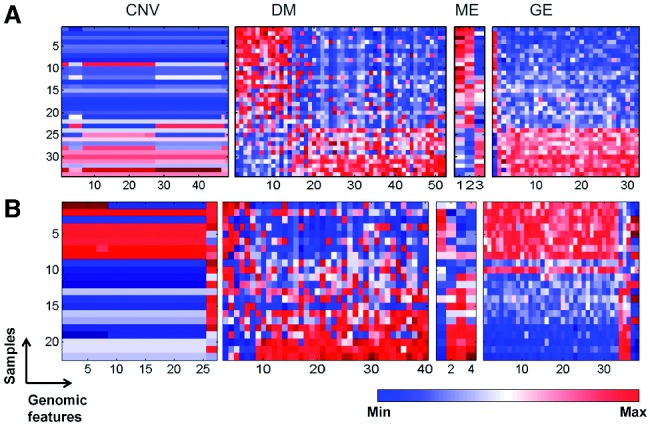
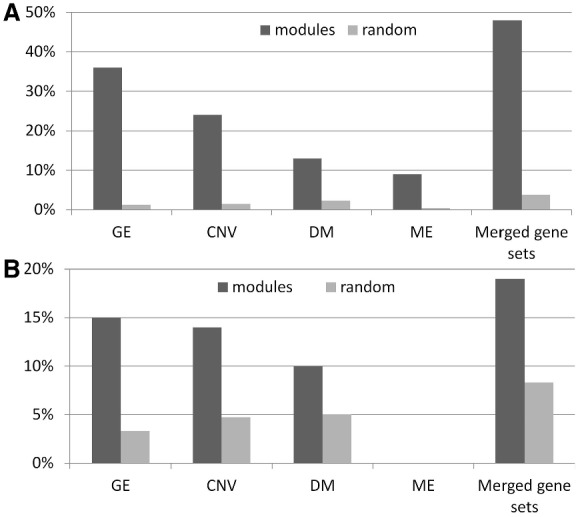
In this article, we introduced a sparse Multi-Block Partial Least Squares (sMBPLS) regression method to identify multi-dimensional regulatory modules from this new type of data. A multi-dimensional regulatory module contains sets of regulatory factors from different layers that are likely to jointly contribute to a local 'gene expression factory'. We demonstrated the performance of our method on the simulated data as well as on The Cancer Genomic Atlas Ovarian Cancer datasets including the CNV, DM, ME and GE data measured on 230 samples. We showed that majority of identified modules have significant functional and transcriptional enrichment, higher than that observed in modules identified using only a single type of genomic data. Our network analysis of the modules revealed that the CNV, DM and microRNA can have coupled impact on expression of important oncogenes and tumor suppressor genes.
The source code implemented by MATLAB is freely available at: http://zhoulab.usc.edu/sMBPLS/.
Supplementary material are available at Bioinformatics online.
真核基因表达(GE)受到精确协调的多层次控制,跨越表观遗传、转录和转录后调控的水平。最近,新兴的多维基因组数据集为研究跨层调控相互作用提供了前所未有的机会。在这些数据集中,同一组样本在几个基因组活动层面上进行了分析,例如拷贝数变异(CNV)、DNA 甲基化(DM)、GE 和 microRNA 表达(ME)。然而,目前适合此类数据的分析方法还很少。
在本文中,我们引入了一种稀疏多块偏最小二乘(sMBPLS)回归方法,用于从这种新型数据中识别多维调控模块。一个多维调控模块包含来自不同层的调控因子集,这些因子可能共同促成局部的“基因表达工厂”。我们在模拟数据以及包括 230 个样本的 CNV、DM、ME 和 GE 数据的癌症基因组图谱卵巢癌数据集上演示了我们方法的性能。我们表明,大多数鉴定的模块具有显著的功能和转录丰度富集,高于仅使用单一类型基因组数据鉴定的模块。我们对模块的网络分析表明,CNV、DM 和 microRNA 可以对重要癌基因和肿瘤抑制基因的表达产生耦合影响。
用 MATLAB 实现的源代码可在以下网址免费获得:http://zhoulab.usc.edu/sMBPLS/。
补充材料可在生物信息学在线获得。