Sydney Medical School, The University of Sydney, Sydney, New South Wales, Australia.
PLoS One. 2011;6(6):e19517. doi: 10.1371/journal.pone.0019517. Epub 2011 Jun 8.
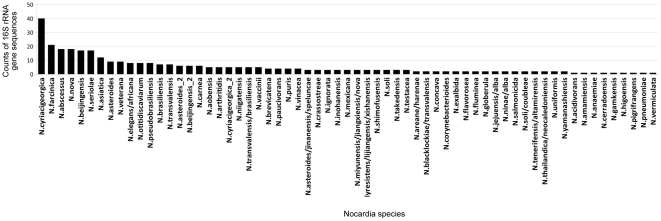
The intra- and inter-species genetic diversity of bacteria and the absence of 'reference', or the most representative, sequences of individual species present a significant challenge for sequence-based identification. The aims of this study were to determine the utility, and compare the performance of several clustering and classification algorithms to identify the species of 364 sequences of 16S rRNA gene with a defined species in GenBank, and 110 sequences of 16S rRNA gene with no defined species, all within the genus Nocardia.
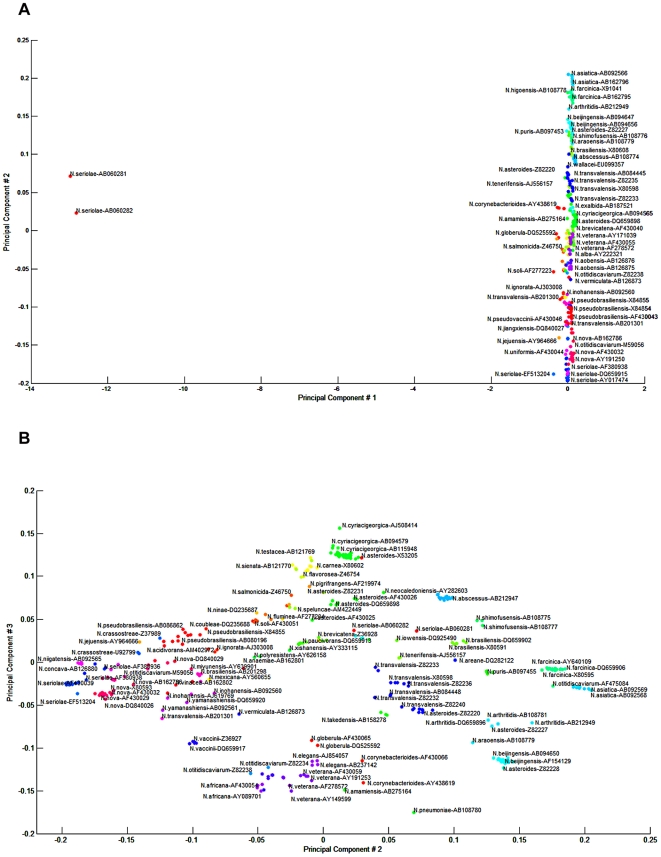
A total of 364 16S rRNA gene sequences of Nocardia species were studied. In addition, 110 16S rRNA gene sequences assigned only to the Nocardia genus level at the time of submission to GenBank were used for machine learning classification experiments. Different clustering algorithms were compared with a novel algorithm or the linear mapping (LM) of the distance matrix. Principal Components Analysis was used for the dimensionality reduction and visualization.
The LM algorithm achieved the highest performance and classified the set of 364 16S rRNA sequences into 80 clusters, the majority of which (83.52%) corresponded with the original species. The most representative 16S rRNA sequences for individual Nocardia species have been identified as 'centroids' in respective clusters from which the distances to all other sequences were minimized; 110 16S rRNA gene sequences with identifications recorded only at the genus level were classified using machine learning methods. Simple kNN machine learning demonstrated the highest performance and classified Nocardia species sequences with an accuracy of 92.7% and a mean frequency of 0.578.
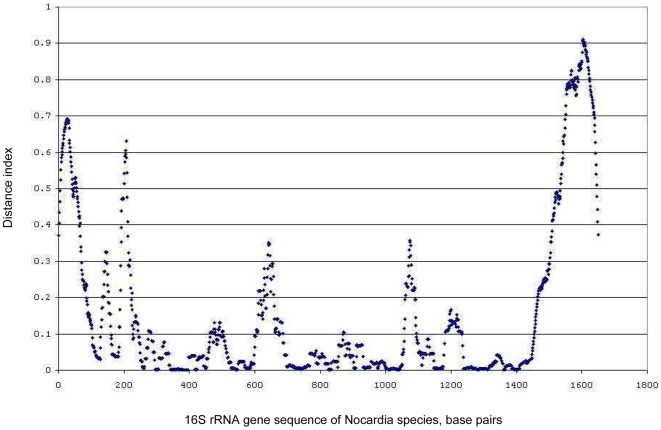
The identification of centroids of 16S rRNA gene sequence clusters using novel distance matrix clustering enables the identification of the most representative sequences for each individual species of Nocardia and allows the quantitation of inter- and intra-species variability.
细菌的种内和种间遗传多样性以及缺乏“参考”(即最具代表性的)单个物种序列,给基于序列的鉴定带来了重大挑战。本研究旨在确定几种聚类和分类算法的效用,并比较它们的性能,以鉴定 GenBank 中定义的种属内 364 条 16S rRNA 基因序列和 110 条无明确种属的 16S rRNA 基因序列的物种,这些序列均属于诺卡氏菌属。
研究了总共 364 条诺卡氏菌属 16S rRNA 基因序列。此外,还使用在提交到 GenBank 时仅被归类为诺卡氏菌属水平的 110 条 16S rRNA 基因序列进行机器学习分类实验。比较了不同的聚类算法与一种新算法或距离矩阵的线性映射(LM)。主成分分析用于降维和可视化。
LM 算法的性能最高,将 364 条 16S rRNA 序列集分为 80 个聚类,其中大多数(83.52%)与原始物种相对应。已确定各个诺卡氏菌属物种的最具代表性的 16S rRNA 序列为各自聚类的“质心”,从这些聚类中可以最小化到所有其他序列的距离;使用机器学习方法对仅在属水平上记录有鉴定的 110 条 16S rRNA 基因序列进行分类。简单的 kNN 机器学习显示出最高的性能,对诺卡氏菌属物种序列的分类准确率为 92.7%,平均频率为 0.578。
使用新的距离矩阵聚类方法确定 16S rRNA 基因序列聚类的质心,可鉴定诺卡氏菌属每个种属的最具代表性的序列,并可量化种间和种内变异性。