Isborn Christine M, Luehr Nathan, Ufimtsev Ivan S, Martínez Todd J
J Chem Theory Comput. 2011 Jun 14;7(6):1814-1823. doi: 10.1021/ct200030k. Epub 2011 May 12.
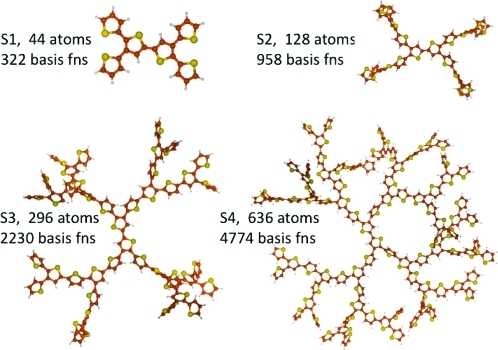
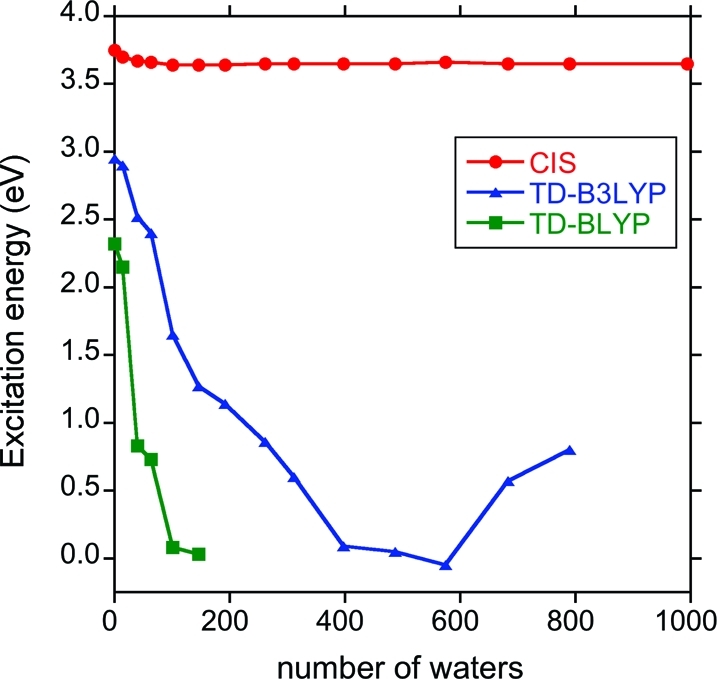
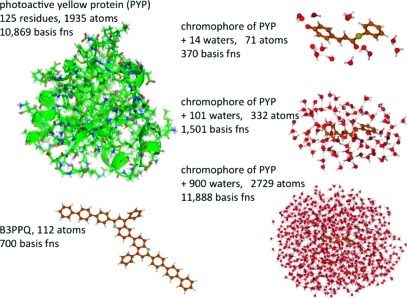
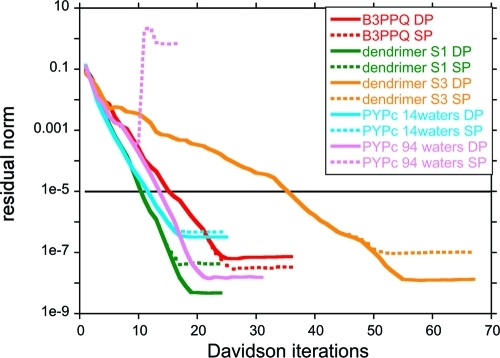
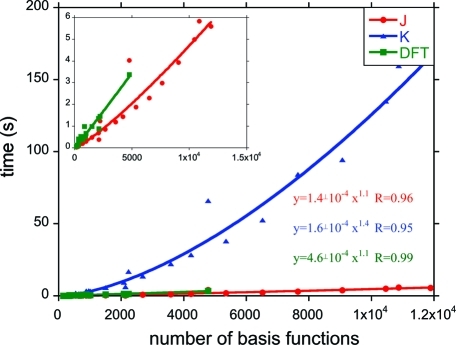
Excited-state calculations are implemented in a development version of the GPU-based TeraChem software package using the configuration interaction singles (CIS) and adiabatic linear response Tamm-Dancoff time-dependent density functional theory (TDA-TDDFT) methods. The speedup of the CIS and TDDFT methods using GPU-based electron repulsion integrals and density functional quadrature integration allows full ab initio excited-state calculations on molecules of unprecedented size. CIS/6-31G and TD-BLYP/6-31G benchmark timings are presented for a range of systems, including four generations of oligothiophene dendrimers, photoactive yellow protein (PYP), and the PYP chromophore solvated with 900 quantum mechanical water molecules. The effects of double and single precision integration are discussed, and mixed precision GPU integration is shown to give extremely good numerical accuracy for both CIS and TDDFT excitation energies (excitation energies within 0.0005 eV of extended double precision CPU results).
基于GPU的TeraChem软件包的开发版本中,使用单激发组态相互作用(CIS)和绝热线性响应Tamm-Dancoff含时密度泛函理论(TDA-TDDFT)方法进行激发态计算。利用基于GPU的电子排斥积分和密度泛函求积积分对CIS和TDDFT方法进行加速,能够对前所未有的大尺寸分子进行全从头算激发态计算。给出了一系列体系的CIS/6-31G和TD-BLYP/6-31G基准计时结果,包括四代寡聚噻吩树枝状大分子、光活性黄色蛋白(PYP)以及用900个量子力学水分子溶剂化的PYP发色团。讨论了双精度积分和单精度积分的影响,结果表明混合精度GPU积分对CIS和TDDFT激发能都具有极高的数值精度(激发能与扩展双精度CPU结果相差在0.0005 eV以内)。