Si Jingna, Zhang Zengming, Lin Biaoyang, Schroeder Michael, Huang Bingding
Systems Biology Division, Zhejiang-California International NanoSystems Institute, Zhejiang University, Kaixuan Road 268, Hangzhou, China.
BMC Syst Biol. 2011 Jun 20;5 Suppl 1(Suppl 1):S7. doi: 10.1186/1752-0509-5-S1-S7.
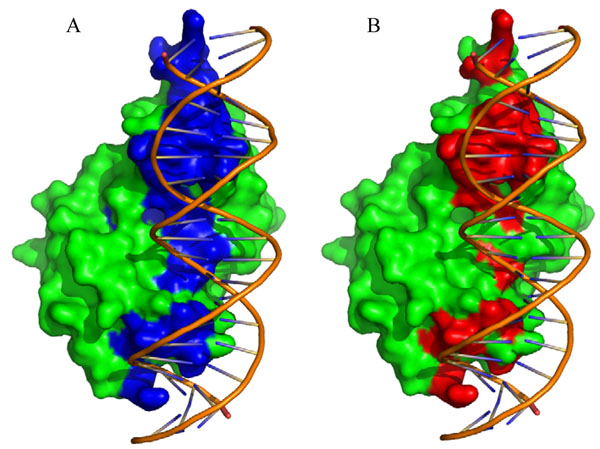
Protein-DNA interactions play an important role in many fundamental biological activities such as DNA replication, transcription and repair. Identification of amino acid residues involved in DNA binding site is critical for understanding of the mechanism of gene regulations. In the last decade, there have been a number of computational approaches developed to predict protein-DNA binding sites based on protein sequence and/or structural information.
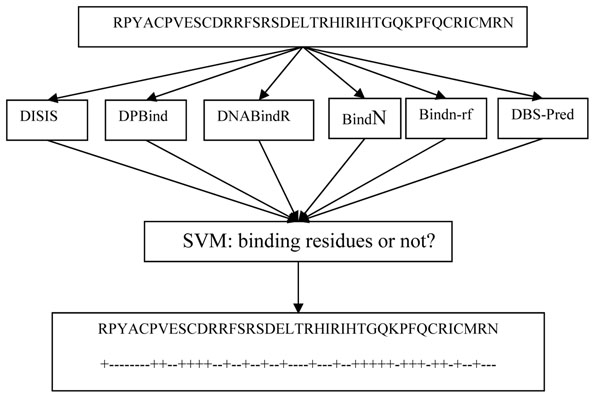
In this article, we present metaDBSite, a meta web server to predict DNA-binding residues for DNA-binding proteins. MetaDBSite integrates the prediction results from six available online web servers: DISIS, DNABindR, BindN, BindN-rf, DP-Bind and DBS-PRED and it solely uses sequence information of proteins. A large dataset of DNA-binding proteins is constructed from the Protein Data Bank and it serves as a gold-standard benchmark to evaluate the metaDBSite approach and the other six predictors.
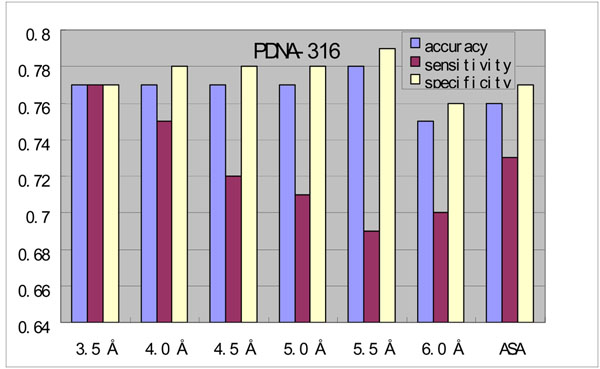
The comparison results show that metaDBSite outperforms single individual approach. We believe that metaDBSite will become a useful and integrative tool for protein DNA-binding residues prediction. The MetaDBSite web-server is freely available at http://projects.biotec.tu-dresden.de/metadbsite/ and http://sysbio.zju.edu.cn/metadbsite.
蛋白质与DNA的相互作用在许多基本生物学活动中发挥着重要作用,如DNA复制、转录和修复。识别参与DNA结合位点的氨基酸残基对于理解基因调控机制至关重要。在过去十年中,已经开发了许多基于蛋白质序列和/或结构信息来预测蛋白质-DNA结合位点的计算方法。
在本文中,我们展示了metaDBSite,一个用于预测DNA结合蛋白的DNA结合残基的元网络服务器。MetaDBSite整合了来自六个可用在线网络服务器(DISIS、DNABindR、BindN、BindN-rf、DP-Bind和DBS-PRED)的预测结果,并且仅使用蛋白质的序列信息。从蛋白质数据库构建了一个大型DNA结合蛋白数据集,它作为评估metaDBSite方法和其他六个预测器的黄金标准基准。
比较结果表明metaDBSite优于单个方法。我们相信metaDBSite将成为预测蛋白质DNA结合残基的一个有用且综合的工具。MetaDBSite网络服务器可在http://projects.biotec.tu-dresden.de/metadbsite/和http://sysbio.zju.edu.cn/metadbsite免费获取。