Bio-Pharmaceutical Key Laboratory of Heilongjiang Province, and College of Bioinformatics Science and Technology, Harbin Medical University, Harbin, China.
PLoS One. 2011;6(6):e21131. doi: 10.1371/journal.pone.0021131. Epub 2011 Jun 17.
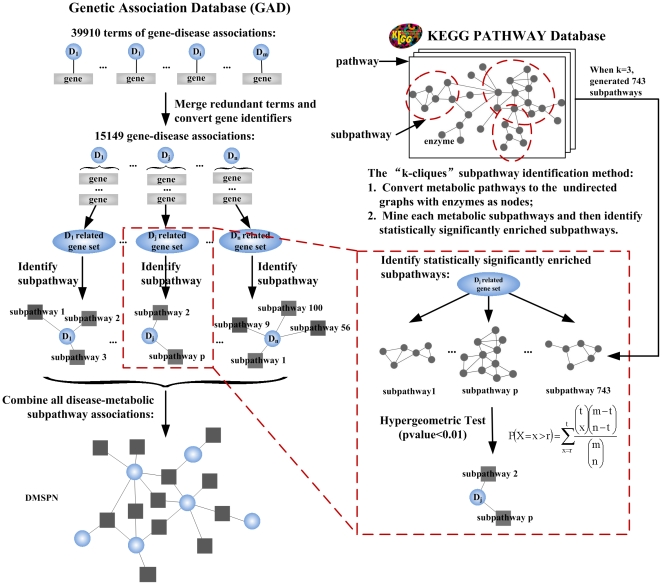
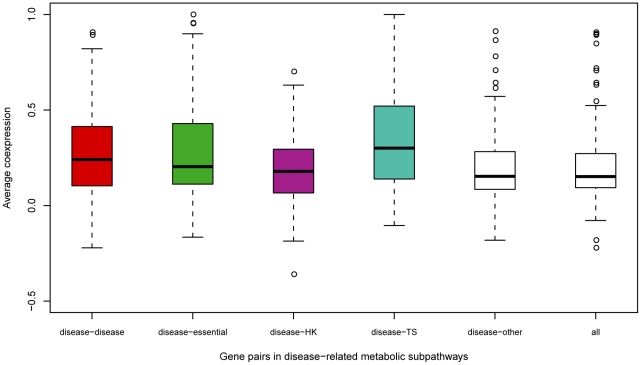
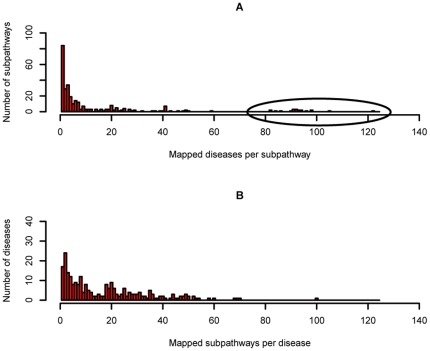
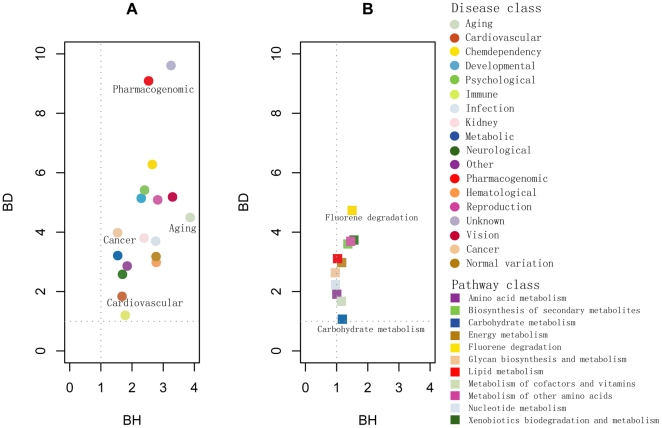
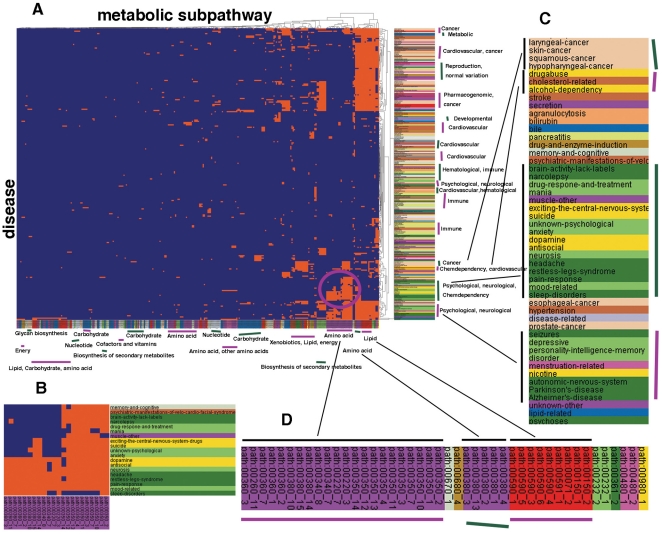
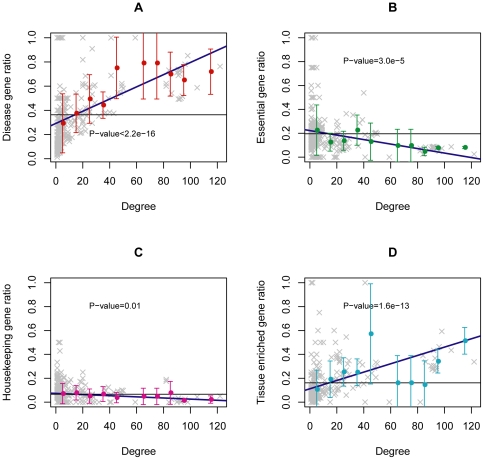
One of the challenging problems in the etiology of diseases is to explore the relationships between initiation and progression of diseases and abnormalities in local regions of metabolic pathways. To gain insight into such relationships, we applied the "k-clique" subpathway identification method to all disease-related gene sets. For each disease, the disease risk regions of metabolic pathways were then identified and considered as subpathways associated with the disease. We finally built a disease-metabolic subpathway network (DMSPN). Through analyses based on network biology, we found that a few subpathways, such as that of cytochrome P450, were highly connected with many diseases, and most belonged to fundamental metabolisms, suggesting that abnormalities of fundamental metabolic processes tend to cause more types of diseases. According to the categories of diseases and subpathways, we tested the clustering phenomenon of diseases and metabolic subpathways in the DMSPN. The results showed that both disease nodes and subpathway nodes displayed slight clustering phenomenon. We also tested correlations between network topology and genes within disease-related metabolic subpathways, and found that within a disease-related subpathway in the DMSPN, the ratio of disease genes and the ratio of tissue-specific genes significantly increased as the number of diseases caused by the subpathway increased. Surprisingly, the ratio of essential genes significantly decreased and the ratio of housekeeping genes remained relatively unchanged. Furthermore, the coexpression levels between disease genes and other types of genes were calculated for each subpathway in the DMSPN. The results indicated that those genes intensely influenced by disease genes, including essential genes and tissue-specific genes, might be significantly associated with the disease diversity of subpathways, suggesting that different kinds of genes within a disease-related subpathway may play significantly differential roles on the diversity of diseases caused by the corresponding subpathway.
疾病病因学中的一个挑战性问题是探索疾病的起始和进展与代谢途径局部区域的异常之间的关系。为了深入了解这种关系,我们将“k 团块”子路径识别方法应用于所有与疾病相关的基因集。然后,确定了每种疾病的代谢途径疾病风险区域,并将其视为与疾病相关的子路径。最后,我们构建了疾病-代谢子路径网络(DMSPN)。通过基于网络生物学的分析,我们发现少数子路径,如细胞色素 P450 途径,与许多疾病高度相关,且大多数属于基础代谢,这表明基础代谢过程的异常更容易导致多种类型的疾病。根据疾病和代谢子路径的类别,我们测试了 DMSPN 中疾病和代谢子路径的聚类现象。结果表明,疾病节点和子路径节点都显示出轻微的聚类现象。我们还测试了网络拓扑和疾病相关代谢子路径内基因之间的相关性,发现 DMSPN 中与疾病相关的子路径内,随着该子路径引起的疾病数量的增加,疾病基因与组织特异性基因的比例显著增加。令人惊讶的是,必需基因的比例显著降低,管家基因的比例保持相对不变。此外,我们还计算了 DMSPN 中每个子路径中疾病基因与其他类型基因之间的共表达水平。结果表明,那些受疾病基因强烈影响的基因,包括必需基因和组织特异性基因,可能与子路径疾病多样性显著相关,这表明疾病相关子路径内的不同类型基因可能在相应子路径引起的疾病多样性中发挥显著不同的作用。