Department of Health Studies, The University of Chicago, Chicago, IL 60637, USA.
BMC Med Genomics. 2011 Jun 23;4:50. doi: 10.1186/1755-8794-4-50.
We performed a genome-wide scan of 27,578 CpG loci covering 14,475 genes to identify differentially methylated loci (DML) in colorectal carcinoma (CRC).
We used Illumina's Infinium methylation assay in paired DNA samples extracted from 24 fresh frozen CRC tissues and their corresponding normal colon tissues from 24 consecutive diagnosed patients at a tertiary medical center.
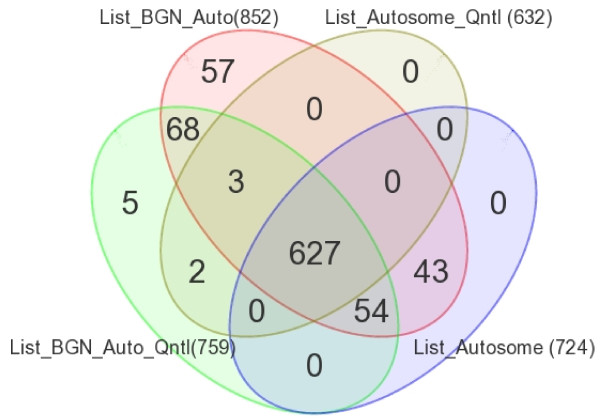
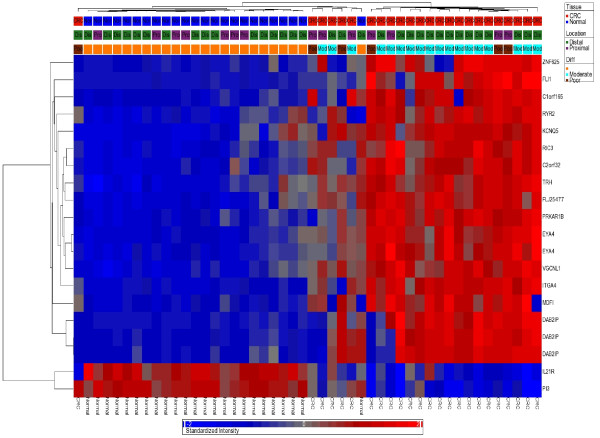
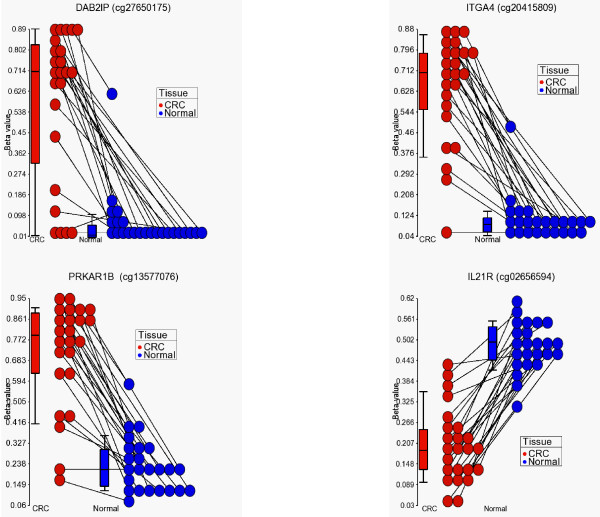
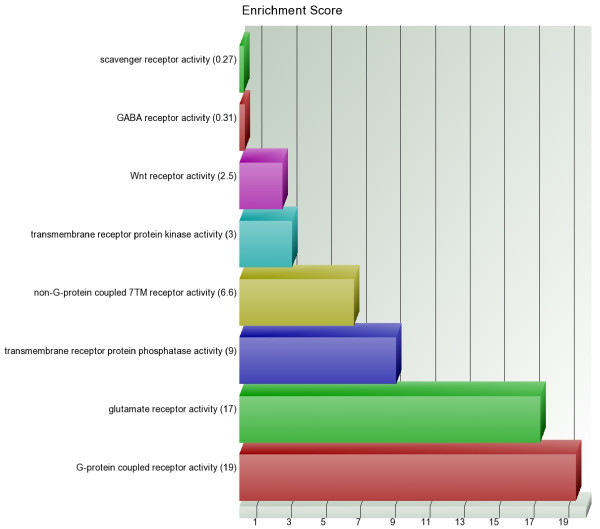
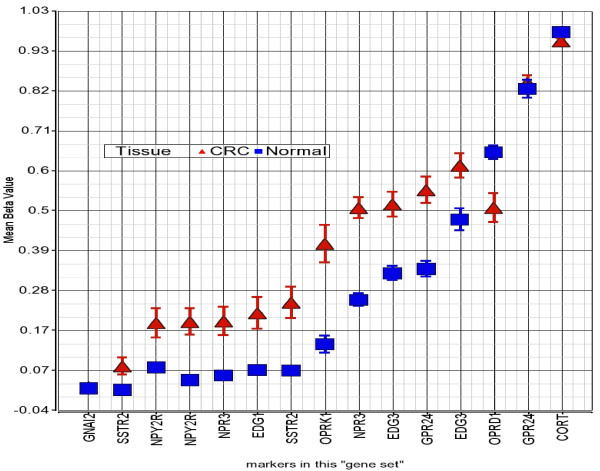
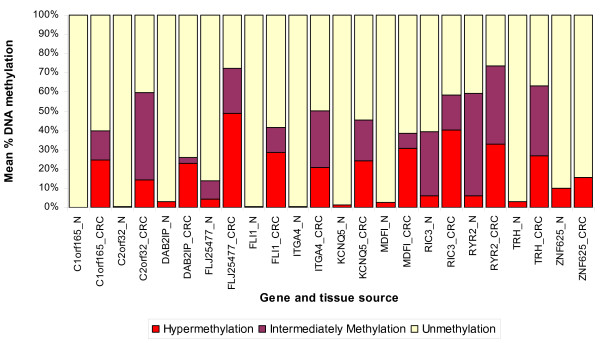
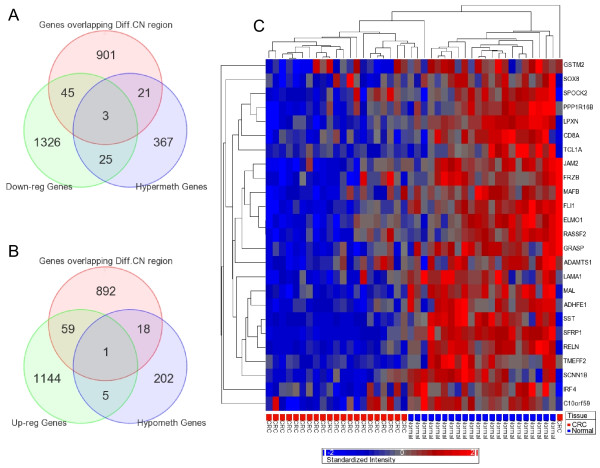
We found a total of 627 DML in CRC covering 513 genes, of which 535 are novel DML covering 465 genes. We also validated the Illumina Infinium methylation data for top-ranking genes by non-bisulfite conversion q-PCR-based methyl profiler assay in a subset of the same samples. We also carried out integration of genome-wide copy number and expression microarray along with methylation profiling to see the functional effect of methylation. Gene Set Enrichment Analysis (GSEA) showed that among the major "gene sets" that are hypermethylated in CRC are the sets: "inhibition of adenylate cyclase activity by G-protein signaling", "Rac guanyl-nucleotide exchange factor activity", "regulation of retinoic acid receptor signaling pathway" and "estrogen receptor activity". Two-level nested cross validation showed that DML-based predictive models may offer reasonable sensitivity (around 89%), specificity (around 95%), positive predictive value (around 95%) and negative predictive value (around 89%), suggesting that these markers may have potential clinical application.
Our genome-wide methylation study in CRC clearly supports most of the previous findings; additionally we found a large number of novel DML in CRC tissue. If confirmed in future studies, these findings may lead to identification of genomic markers for potential clinical application.
我们对 27578 个 CpG 位点(涵盖 14475 个基因)进行了全基因组扫描,以鉴定结直肠癌(CRC)中差异甲基化的位点(DML)。
我们使用 Illumina 的 Infinium 甲基化检测试剂盒,对来自一家三级医疗中心的 24 名连续确诊患者的 24 对新鲜冷冻 CRC 组织及其相应的正常结肠组织的 DNA 样本进行了检测。
我们在 CRC 中发现了总共 627 个 DML,涵盖了 513 个基因,其中 535 个是新的 DML,涵盖了 465 个基因。我们还通过非亚硫酸氢盐转化 qPCR 甲基化分析验证了同一批样本中排名靠前的基因的 Illumina Infinium 甲基化数据。我们还进行了全基因组拷贝数和表达微阵列与甲基化谱的整合,以观察甲基化的功能影响。基因集富集分析(GSEA)表明,在 CRC 中高度甲基化的主要“基因集”包括:“G 蛋白信号抑制腺苷酸环化酶活性”、“Rac 鸟苷酸交换因子活性”、“视黄酸受体信号通路的调节”和“雌激素受体活性”。两级嵌套交叉验证表明,基于 DML 的预测模型可能具有合理的敏感性(约 89%)、特异性(约 95%)、阳性预测值(约 95%)和阴性预测值(约 89%),这表明这些标志物可能具有潜在的临床应用价值。
我们在 CRC 中的全基因组甲基化研究清楚地支持了大多数先前的发现;此外,我们还在 CRC 组织中发现了大量新的 DML。如果在未来的研究中得到证实,这些发现可能会导致潜在临床应用的基因组标记的识别。