Department of Entomology, University of Maryland, College Park, MD, USA.
BMC Evol Biol. 2011 Jun 24;11:182. doi: 10.1186/1471-2148-11-182.
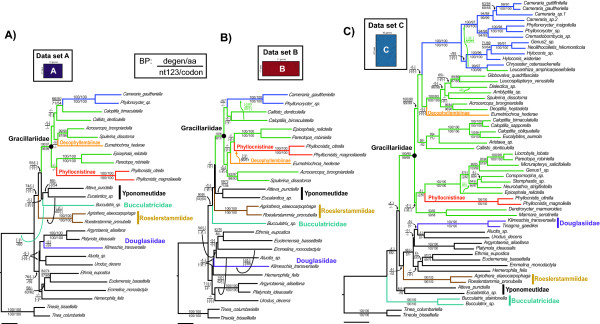
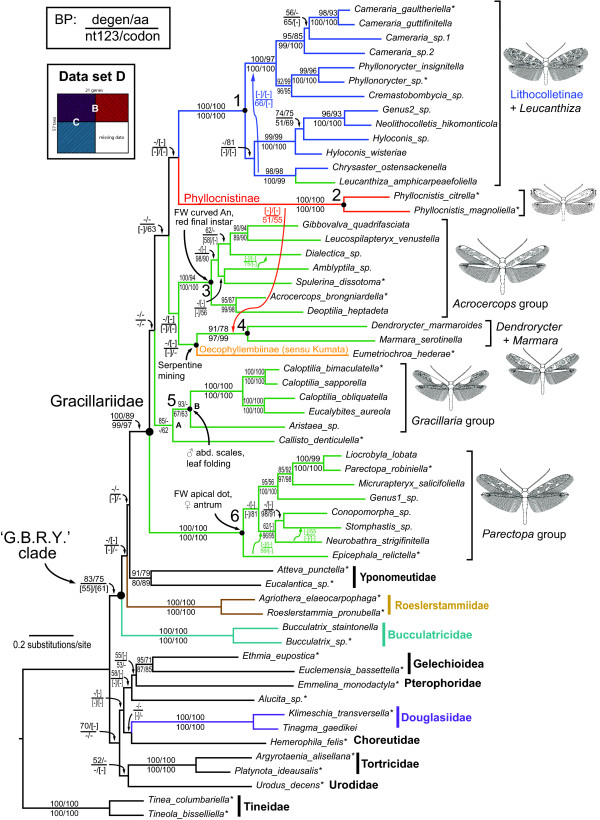
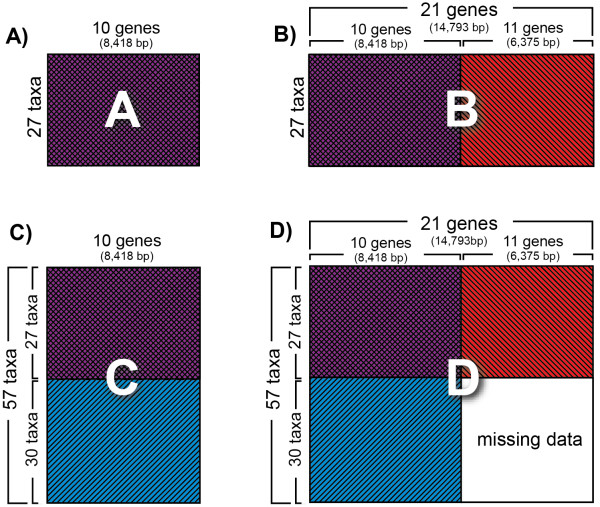
Researchers conducting molecular phylogenetic studies are frequently faced with the decision of what to do when weak branch support is obtained for key nodes of importance. As one solution, the researcher may choose to sequence additional orthologous genes of appropriate evolutionary rate for the taxa in the study. However, generating large, complete data matrices can become increasingly difficult as the number of characters increases. A few empirical studies have shown that augmenting genes even for a subset of taxa can improve branch support. However, because each study differs in the number of characters and taxa, there is still a need for additional studies that examine whether incomplete sampling designs are likely to aid at increasing deep node resolution. We target Gracillariidae, a Cretaceous-age (~100 Ma) group of leaf-mining moths to test whether the strategy of adding genes for a subset of taxa can improve branch support for deep nodes. We initially sequenced ten genes (8,418 bp) for 57 taxa that represent the major lineages of Gracillariidae plus outgroups. After finding that many deep divergences remained weakly supported, we sequenced eleven additional genes (6,375 bp) for a 27-taxon subset. We then compared results from different data sets to assess whether one sampling design can be favored over another. The concatenated data set comprising all genes and all taxa and three other data sets of different taxon and gene sub-sampling design were analyzed with maximum likelihood. Each data set was subject to five different models and partitioning schemes of non-synonymous and synonymous changes. Statistical significance of non-monophyly was examined with the Approximately Unbiased (AU) test.
Partial augmentation of genes led to high support for deep divergences, especially when non-synonymous changes were analyzed alone. Increasing the number of taxa without an increase in number of characters led to lower bootstrap support; increasing the number of characters without increasing the number of taxa generally increased bootstrap support. More than three-quarters of nodes were supported with bootstrap values greater than 80% when all taxa and genes were combined. Gracillariidae, Lithocolletinae + Leucanthiza, and Acrocercops and Parectopa groups were strongly supported in nearly every analysis. Gracillaria group was well supported in some analyses, but less so in others. We find strong evidence for the exclusion of Douglasiidae from Gracillarioidea sensu Davis and Robinson (1998). Our results strongly support the monophyly of a G.B.R.Y. clade, a group comprised of Gracillariidae + Bucculatricidae + Roeslerstammiidae + Yponomeutidae, when analyzed with non-synonymous changes only, but this group was frequently split when synonymous and non-synonymous substitutions were analyzed together.
进行分子系统发育研究的研究人员经常面临这样的决策,即在获得重要关键节点的弱分支支持时该怎么办。作为一种解决方案,研究人员可以选择为研究中的分类群序列具有适当进化率的额外同源基因。然而,随着字符数量的增加,生成大型完整数据矩阵会变得越来越困难。一些经验研究表明,即使对于分类群的子集增加基因也可以提高分支支持。然而,由于每个研究在字符和分类群的数量上都有所不同,因此仍然需要更多的研究来检查不完全采样设计是否有可能有助于增加深节点分辨率。我们以 Gracillariidae 为目标,这是一组白垩纪(约 100 Ma)的叶潜蛾,以测试为分类群的子集增加基因的策略是否可以提高深节点的分支支持。我们最初为代表 Gracillariidae 主要谱系的 57 个分类群和外群序列了十个基因(8418 bp)。在发现许多深分歧仍然支持较弱后,我们为 27 个分类群子集序列了十一个额外基因(6375 bp)。然后,我们比较了不同数据集的结果,以评估一种采样设计是否可以优于另一种。包含所有基因和所有分类群的串联数据集和三个不同分类群和基因子采样设计的其他三个数据集与最大似然法进行了分析。每个数据集都经过了五个不同的模型和非同义与同义变化的分区方案。使用近似无偏(AU)检验检查非单系性的统计显着性。
部分增加基因导致深分歧的高支持,尤其是当单独分析非同义变化时。增加分类群的数量而不增加字符数量会导致较低的引导支持; 增加字符数量而不增加分类群的数量通常会增加引导支持。当所有分类群和基因组合在一起时,超过四分之三的节点得到了大于 80%的bootstrap 值支持。Gracillariidae、Lithocolletinae + Leucanthiza 和 Acrocercops 和 Parectopa 组在几乎所有分析中都得到了强烈支持。Gracillaria 组在某些分析中得到了很好的支持,但在其他分析中则不然。我们发现有强有力的证据表明将 Douglasiidae 排除在 Davis 和 Robinson(1998)定义的 Gracillarioidea 之外。当仅分析非同义变化时,我们的结果强烈支持 G.B.R.Y. 进化枝的单系性,该进化枝由 Gracillariidae + Bucculatricidae + Roeslerstammiidae + Yponomeutidae 组成,但当同时分析同义和非同义替换时,该进化枝经常分裂。
1)部分或完全增加具有更多字符的数据集可以增加特定深节点的引导支持,并且当单独分析非同义变化时,这种增加是巨大的。因此,添加具有低水平饱和和组成异质性的位点可以大大改善结果。2)Davis 和 Robinson(1998)定义的 Gracillarioidea 显然不包括 Douglasiidae,因此需要对当前的分类进行修改。3)Gracillariidae 在所有进行的分析中都是单系的,尽管这些分类群之间的关系仍不清楚,但几乎所有的物种都可以归入六个强烈支持的进化枝中。4)Bucculatricidae 系统发育位置难以确定的原因可能归因于第三密码子位置的组成异质性。从我们对组成异质性的测试和当排除同义变化时获得的强烈引导值,我们暂时得出结论,Bucculatricidae 与 Gracillariidae + Roeslerstammiidae + Yponomeutidae 密切相关。