Department of Biochemistry and Molecular Biophysics, California Institute of Technology, Pasadena, CA 91125, USA.
Science. 2011 Jul 1;333(6038):58-62. doi: 10.1126/science.1200758.
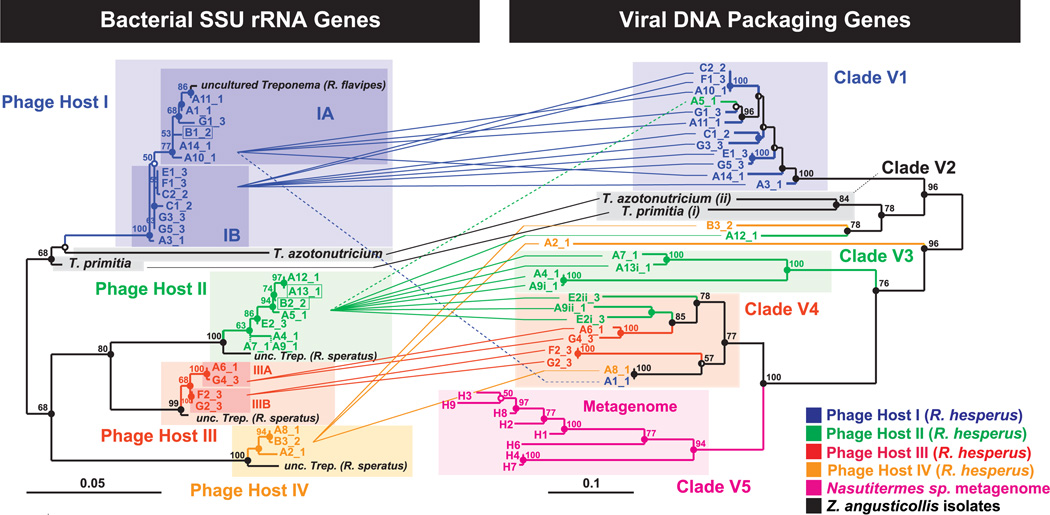
Viruses may very well be the most abundant biological entities on the planet. Yet neither metagenomic studies nor classical phage isolation techniques have shed much light on the identity of the hosts of most viruses. We used a microfluidic digital polymerase chain reaction (PCR) approach to physically link single bacterial cells harvested from a natural environment with a viral marker gene. When we implemented this technique on the microbial community residing in the termite hindgut, we found genus-wide infection patterns displaying remarkable intragenus selectivity. Viral marker allelic diversity revealed restricted mixing of alleles between hosts, indicating limited lateral gene transfer of these alleles despite host proximity. Our approach does not require culturing hosts or viruses and provides a method for examining virus-bacterium interactions in many environments.
病毒很可能是地球上数量最多的生物实体。然而,无论是宏基因组研究还是经典的噬菌体分离技术,都未能揭示大多数病毒宿主的身份。我们使用微流控数字聚合酶链反应(PCR)方法将从自然环境中收获的单个细菌细胞与病毒标记基因物理连接起来。当我们将这项技术应用于白蚁后肠中存在的微生物群落时,发现了具有广泛属感染模式的高度属内选择性。病毒标记等位基因多样性揭示了宿主之间等位基因的有限混合,表明尽管宿主接近,但这些等位基因的水平基因转移有限。我们的方法不需要培养宿主或病毒,为在许多环境中研究病毒-细菌相互作用提供了一种方法。