HRH Prince Alwaleed Bin Talal Bin Abdulaziz Alsaud Institute for Computational Biomedicine, Weill Cornell Medical College, 1305 York Avenue, New York, NY 10021, USA.
BMC Bioinformatics. 2011 Jul 7;12:277. doi: 10.1186/1471-2105-12-277.
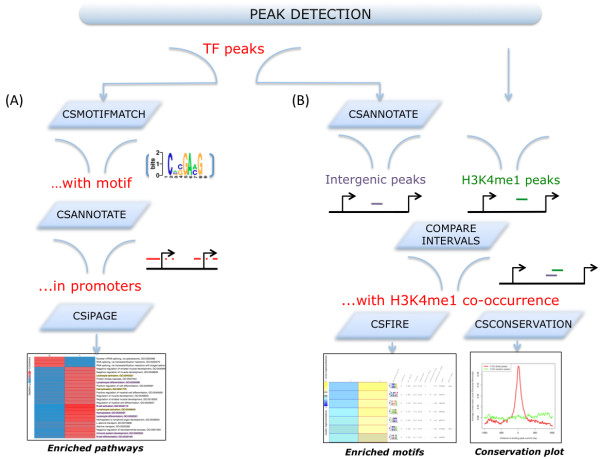
Chromatin immunoprecipitation followed by next generation sequencing (ChIP-seq), enables unbiased and genome-wide mapping of protein-DNA interactions and epigenetic marks. The first step in ChIP-seq data analysis involves the identification of peaks (i.e., genomic locations with high density of mapped sequence reads). The next step consists of interpreting the biological meaning of the peaks through their association with known genes, pathways, regulatory elements, and integration with other experiments. Although several programs have been published for the analysis of ChIP-seq data, they often focus on the peak detection step and are usually not well suited for thorough, integrative analysis of the detected peaks.
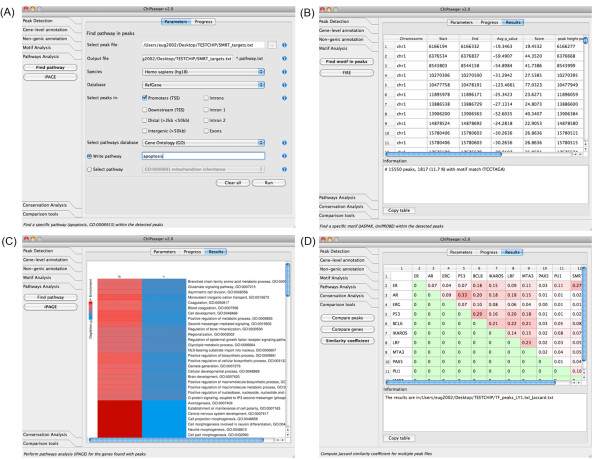
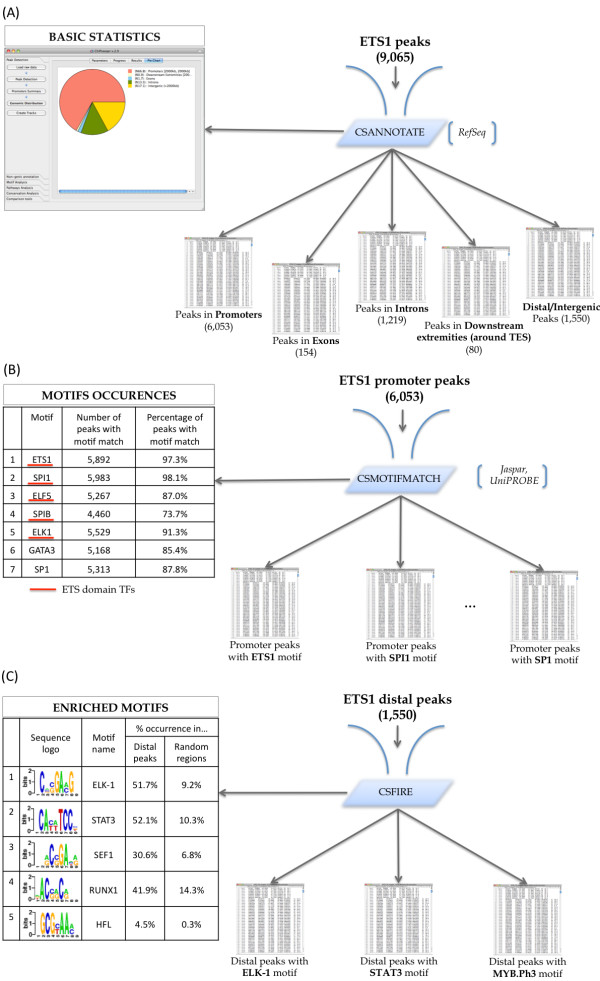
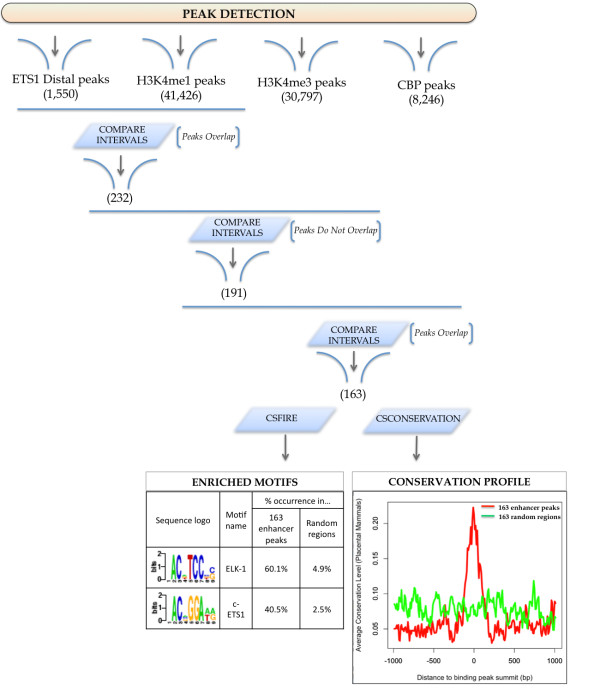
To address the peak interpretation challenge, we have developed ChIPseeqer, an integrative, comprehensive, fast and user-friendly computational framework for in-depth analysis of ChIP-seq datasets. The novelty of our approach is the capability to combine several computational tools in order to create easily customized workflows that can be adapted to the user's needs and objectives. In this paper, we describe the main components of the ChIPseeqer framework, and also demonstrate the utility and diversity of the analyses offered, by analyzing a published ChIP-seq dataset.
ChIPseeqer facilitates ChIP-seq data analysis by offering a flexible and powerful set of computational tools that can be used in combination with one another. The framework is freely available as a user-friendly GUI application, but all programs are also executable from the command line, thus providing flexibility and automatability for advanced users.
染色质免疫沉淀结合下一代测序(ChIP-seq)可实现蛋白质-DNA 相互作用和表观遗传标记的无偏和全基因组作图。ChIP-seq 数据分析的第一步涉及鉴定峰(即具有高密度映射序列读数的基因组位置)。下一步包括通过与已知基因、途径、调控元件的关联以及与其他实验的整合来解释峰的生物学意义。尽管已经发布了用于分析 ChIP-seq 数据的多个程序,但它们通常侧重于峰检测步骤,并且通常不适合对检测到的峰进行全面、综合的分析。
为了解决峰解释的挑战,我们开发了 ChIPseeqer,这是一个用于深入分析 ChIP-seq 数据集的集成、全面、快速且用户友好的计算框架。我们方法的新颖之处在于能够组合几个计算工具,以便创建可定制的工作流程,以适应用户的需求和目标。在本文中,我们描述了 ChIPseeqer 框架的主要组件,并通过分析已发表的 ChIP-seq 数据集演示了所提供分析的实用性和多样性。
ChIPseeqer 通过提供一组灵活且强大的计算工具来简化 ChIP-seq 数据分析,这些工具可以相互组合使用。该框架作为一个用户友好的 GUI 应用程序免费提供,但所有程序也可以从命令行执行,从而为高级用户提供了灵活性和自动化。