IFM Bioinformatics and SeRC (Swedish e-Science Research Centre), Linköping University, S-581 83 Linköping, Sweden.
BMC Bioinformatics. 2011 Jul 19;12:293. doi: 10.1186/1471-2105-12-293.
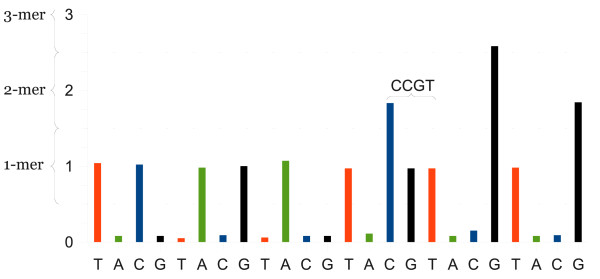
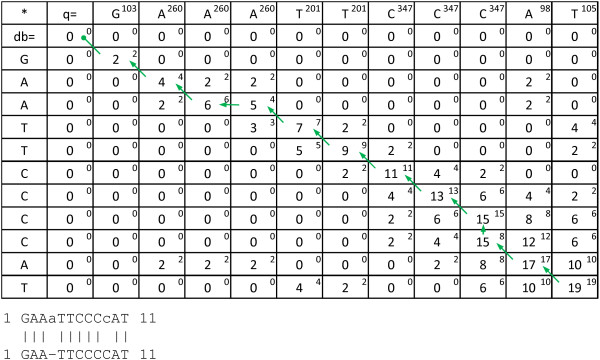
High throughput pyrosequencing (454 sequencing) is the major sequencing platform for producing long read high throughput data. While most other sequencing techniques produce reading errors mainly comparable with substitutions, pyrosequencing produce errors mainly comparable with gaps. These errors are less efficiently detected by most conventional alignment programs and may produce inaccurate alignments.
We suggest a novel algorithm for calculating the optimal local alignment which utilises flowpeak information in order to improve alignment accuracy. Flowpeak information can be retained from a 454 sequencing run through interpretation of the binary SFF-file format. This novel algorithm has been implemented in a program named FAAST (Flow-space Assisted Alignment Search Tool).
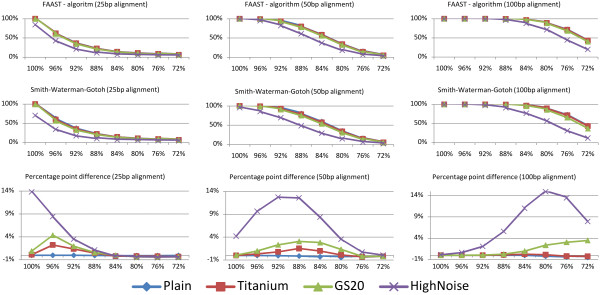
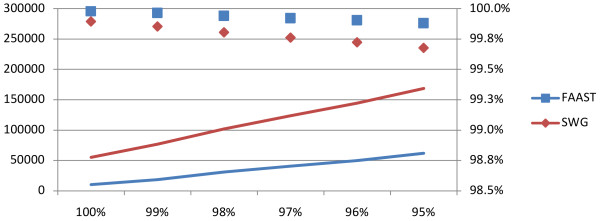
We present and discuss the results of simulations that show that FAAST, through the use of the novel algorithm, can gain several percentage points of accuracy compared to Smith-Waterman-Gotoh alignments, depending on the 454 data quality. Furthermore, through an efficient multi-thread aware implementation, FAAST is able to perform these high quality alignments at high speed. The tool is available at http://www.ifm.liu.se/bioinfo/
高通量焦磷酸测序(454 测序)是产生长读高通量数据的主要测序平台。虽然大多数其他测序技术产生的阅读错误主要与替换相当,但焦磷酸测序产生的错误主要与间隙相当。这些错误在大多数常规对齐程序中检测效率较低,可能会产生不准确的对齐。
我们建议了一种新的算法,用于计算最优局部对齐,该算法利用流峰信息来提高对齐精度。可以通过解释二进制 SFF 文件格式来从 454 测序运行中保留流峰信息。这种新算法已在名为 FAAST(Flow-space Assisted Alignment Search Tool)的程序中实现。
我们展示并讨论了模拟结果,表明 FAAST 通过使用新算法,可以比 Smith-Waterman-Gotoh 对齐获得几个百分点的准确性,具体取决于 454 数据质量。此外,通过高效的多线程感知实现,FAAST 能够以高速进行这些高质量的对齐。该工具可在 http://www.ifm.liu.se/bioinfo/ 获得。