Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, the University of Tokyo, Kashiwa City, Chiba, Japan.
BMC Bioinformatics. 2018 Feb 19;19(Suppl 1):45. doi: 10.1186/s12859-018-2014-8.
The read length of single-molecule DNA sequencers is reaching 1 Mb. Popular alignment software tools widely used for analyzing such long reads often take advantage of single-instruction multiple-data (SIMD) operations to accelerate calculation of dynamic programming (DP) matrices in the Smith-Waterman-Gotoh (SWG) algorithm with a fixed alignment start position at the origin. Nonetheless, 16-bit or 32-bit integers are necessary for storing the values in a DP matrix when sequences to be aligned are long; this situation hampers the use of the full SIMD width of modern processors.
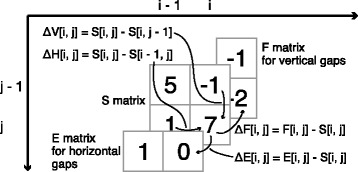
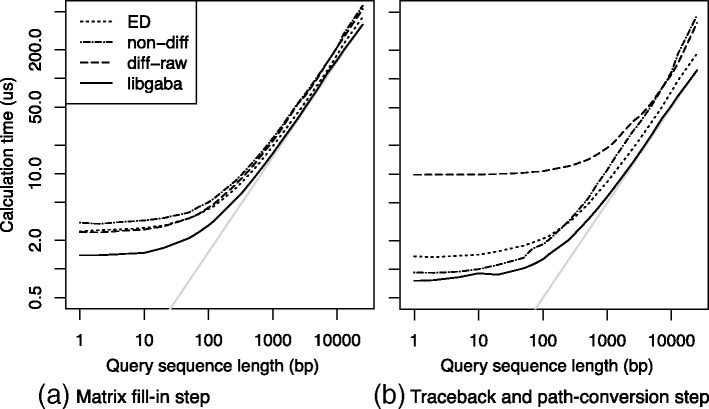
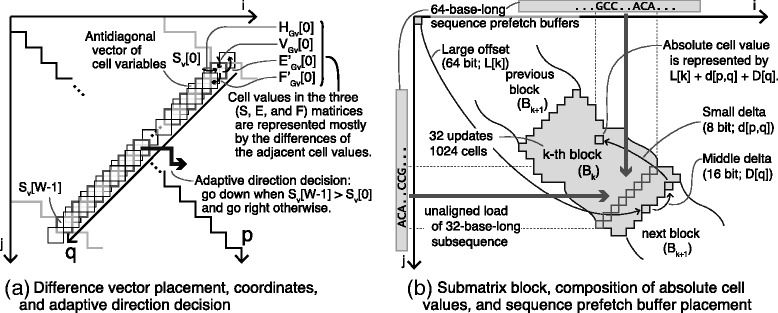
We proposed a faster semi-global alignment algorithm, "difference recurrence relations," that runs more rapidly than the state-of-the-art algorithm by a factor of 2.1. Instead of calculating and storing all the values in a DP matrix directly, our algorithm computes and stores mainly the differences between the values of adjacent cells in the matrix. Although the SWG algorithm and our algorithm can output exactly the same result, our algorithm mainly involves 8-bit integer operations, enabling us to exploit the full width of SIMD operations (e.g., 32) on modern processors. We also developed a library, libgaba, so that developers can easily integrate our algorithm into alignment programs.
Our novel algorithm and optimized library implementation will facilitate accelerating nucleotide long-read analysis algorithms that use pairwise alignment stages. The library is implemented in the C programming language and available at https://github.com/ocxtal/libgaba .
单分子 DNA 测序仪的读长已达到 1Mb。目前广泛应用于分析此类长读长的流行对齐软件工具通常利用单指令多数据(SIMD)操作,以加快在原点固定对齐起始位置的 Smith-Waterman-Gotoh(SWG)算法中的动态规划(DP)矩阵的计算。然而,当待对齐的序列较长时,用于存储 DP 矩阵中值的 16 位或 32 位整数会阻碍现代处理器充分利用其 SIMD 宽度。
我们提出了一种更快的半全局对齐算法“差值递归关系”,其运行速度比最先进的算法快 2.1 倍。与直接计算和存储 DP 矩阵中的所有值不同,我们的算法主要计算和存储矩阵中相邻单元格值之间的差异。虽然 SWG 算法和我们的算法可以输出完全相同的结果,但我们的算法主要涉及 8 位整数运算,使我们能够充分利用现代处理器的 SIMD 操作全宽(例如 32)。我们还开发了一个库 libgaba,以便开发人员可以轻松地将我们的算法集成到对齐程序中。
我们的新颖算法和优化的库实现将有助于加速使用成对对齐阶段的核苷酸长读分析算法。该库是用 C 编程语言实现的,并可在 https://github.com/ocxtal/libgaba 上获得。