Laboratory of Cellular and Molecular Basis of Diseases, Division of Translational Medicine, Wadsworth Center, New York State Department of Health, Albany, New York, United States of America.
PLoS One. 2011;6(7):e21871. doi: 10.1371/journal.pone.0021871. Epub 2011 Jul 11.
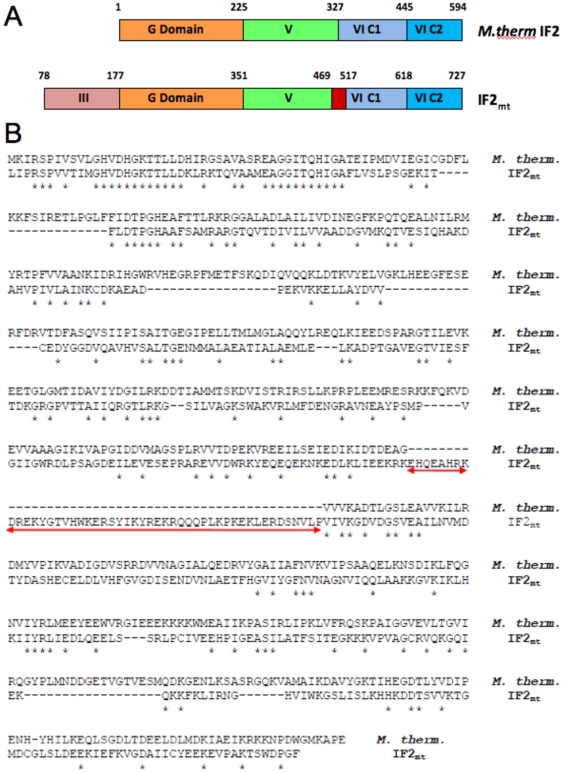
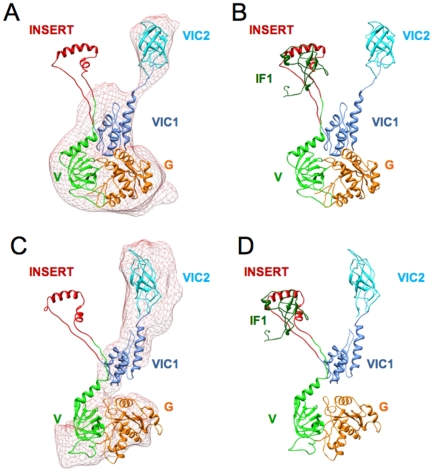
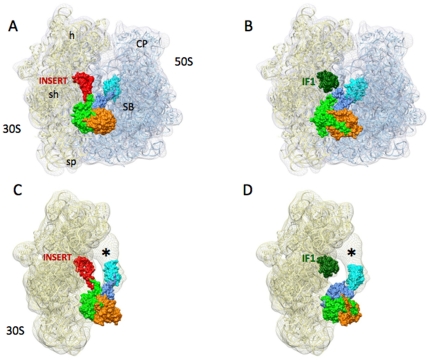
Proteins involved in mammalian mitochondrial translation, when compared to analogous bacterial proteins, frequently have additional sequence regions whose structural or functional roles are not always clear. For example, an additional short insert sequence in the bovine mitochondrial initiation factor 2 (IF2(mt)) seems sufficient to fulfill the added role of eubacterial initiation factor IF1. Prior to our recent cryo-EM study that showed IF2(mt) to structurally occupy both the IF1 and IF2 binding sites, the spatial separation of these sites, and the short length of the insert sequence, posed ambiguity in whether it could perform the role of IF1 through occupation of the IF1 binding site on the ribosome.
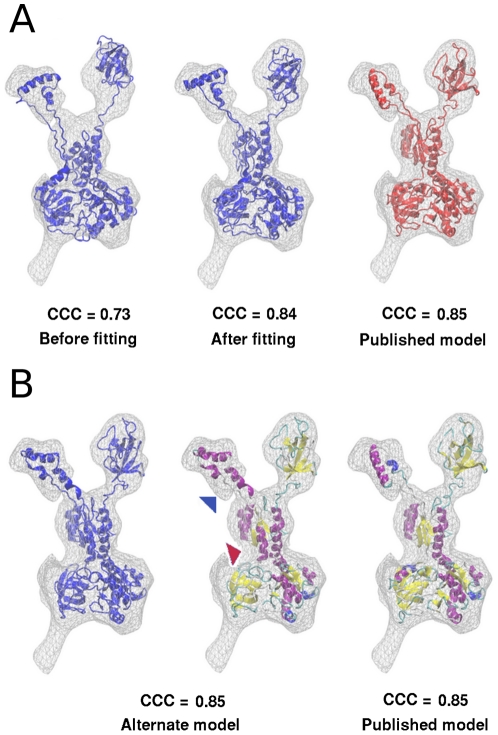
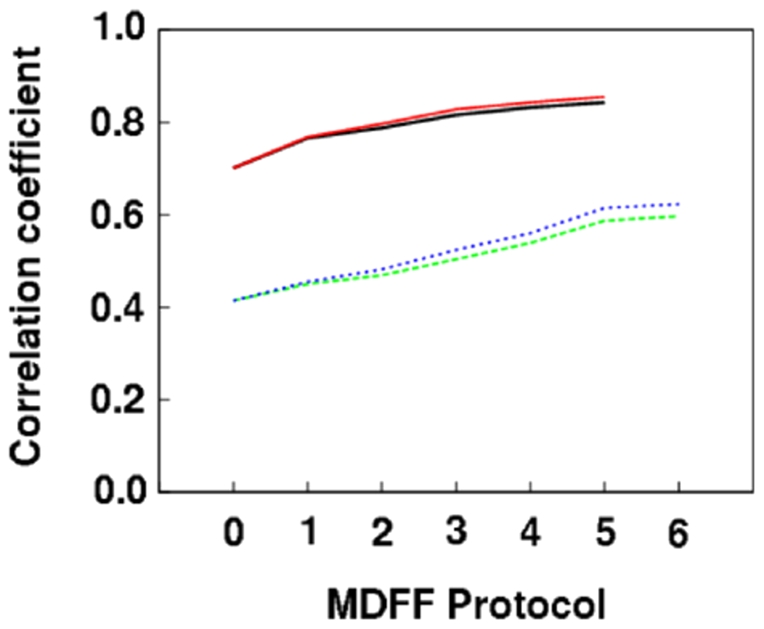
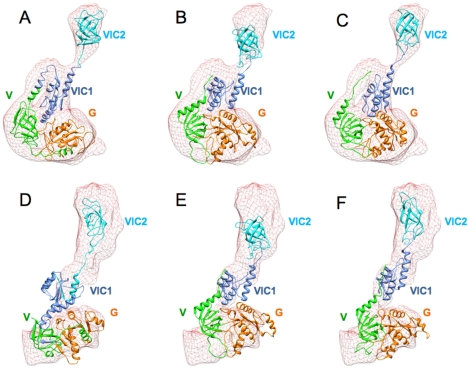
The present study probes how well computational structure prediction methods can a priori address hypothesized roles of such additional sequences by creating quasi-atomic models of IF2(mt) using bacterial IF2 cryo-EM densities (that lack the insert sequences). How such initial IF2(mt) predictions differ from the observed IF2(mt) cryo-EM map and how they can be suitably improved using further sequence analysis and flexible fitting are analyzed.
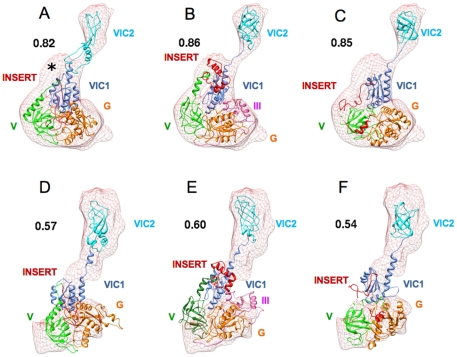
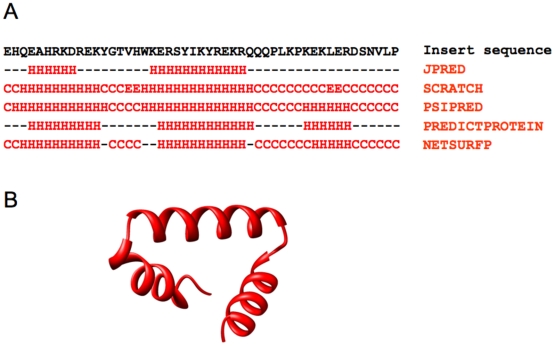
By hypothesizing that the insert sequence occupies the IF1 binding site, continuous IF2(mt) models that occupy both the IF2 and IF1 binding sites can be predicted computationally. These models can be improved by flexible fitting into the IF2(mt) cryo-EM map to get reasonable quasi-atomic IF2(mt) models, but the exact orientation of the insert structure may not be reproduced. Specific eukaryotic insert sequence conservation characteristics can be used to predict alternate IF2(mt) models that have minor secondary structure rearrangements but fewer unusually extended linker regions. Computational structure prediction methods can thus be combined with medium-resolution cryo-EM maps to explore structure-function hypotheses for additional sequence regions and to guide further biochemical experiments, especially in mammalian systems where high-resolution structures are difficult to determine.
与类似的细菌蛋白相比,参与哺乳动物线粒体翻译的蛋白质通常具有额外的序列区域,其结构或功能作用并不总是清楚。例如,牛线粒体起始因子 2(IF2(mt))中的一个额外的短插入序列似乎足以满足真核细菌起始因子 IF1 的附加作用。在我们最近的冷冻电镜研究表明 IF2(mt) 结构上占据 IF1 和 IF2 结合位点之前,这些位点的空间分离以及插入序列的短长度,使得对于它是否可以通过占据核糖体上的 IF1 结合位点来发挥 IF1 的作用存在歧义。
本研究通过使用细菌 IF2 冷冻电镜密度(缺乏插入序列)创建 IF2(mt) 的准原子模型,探讨了计算结构预测方法如何能够预先解决此类额外序列的假设作用。分析了这些初始 IF2(mt) 预测与观察到的 IF2(mt) 冷冻电镜图的差异,以及如何通过进一步的序列分析和灵活拟合来适当改进它们。
通过假设插入序列占据 IF1 结合位点,可以计算出占据 IF2 和 IF1 结合位点的连续 IF2(mt) 模型。通过将这些模型灵活拟合到 IF2(mt) 冷冻电镜图中,可以得到合理的准原子 IF2(mt) 模型,但插入结构的精确方向可能无法重现。特定的真核插入序列保守特征可用于预测具有较小二级结构重排但较少异常扩展连接区的替代 IF2(mt) 模型。因此,计算结构预测方法可以与中分辨率冷冻电镜图结合使用,以探索额外序列区域的结构-功能假设,并指导进一步的生化实验,特别是在难以确定高分辨率结构的哺乳动物系统中。