Center for non-coding RNA in Technology and Health, IBHV University of Copenhagen, Frederiksberg, Denmark.
PLoS Comput Biol. 2011 Aug;7(8):e1002100. doi: 10.1371/journal.pcbi.1002100. Epub 2011 Aug 4.
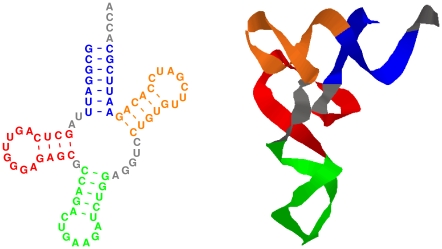
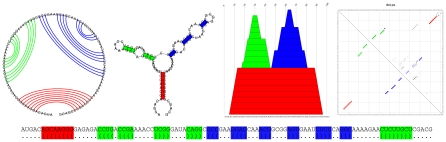
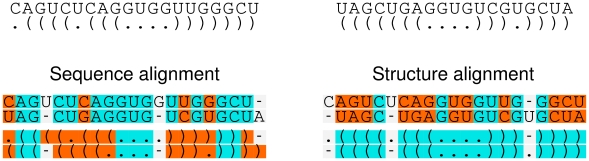
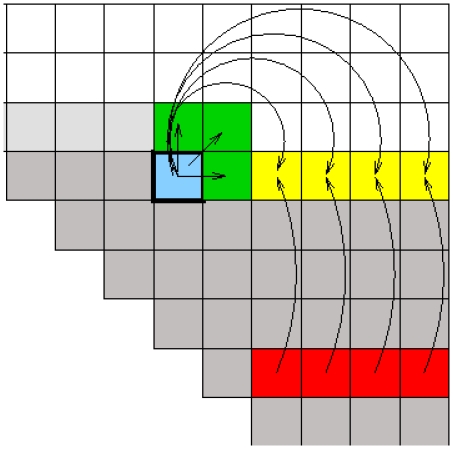
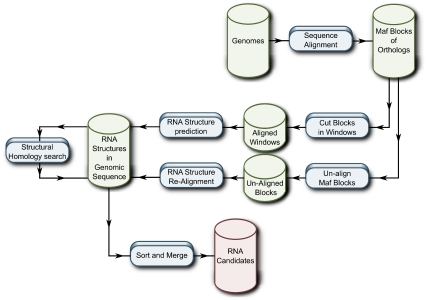
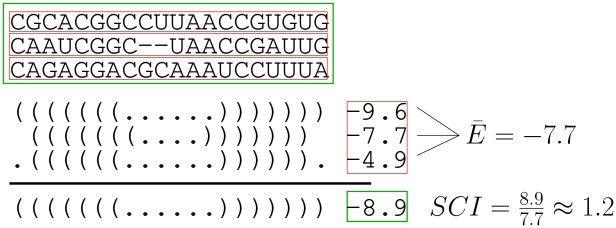
Non-coding RNAs (ncRNAs) are receiving more and more attention not only as an abundant class of genes, but also as regulatory structural elements (some located in mRNAs). A key feature of RNA function is its structure. Computational methods were developed early for folding and prediction of RNA structure with the aim of assisting in functional analysis. With the discovery of more and more ncRNAs, it has become clear that a large fraction of these are highly structured. Interestingly, a large part of the structure is comprised of regular Watson-Crick and GU wobble base pairs. This and the increased amount of available genomes have made it possible to employ structure-based methods for genomic screens. The field has moved from folding prediction of single sequences to computational screens for ncRNAs in genomic sequence using the RNA structure as the main characteristic feature. Whereas early methods focused on energy-directed folding of single sequences, comparative analysis based on structure preserving changes of base pairs has been efficient in improving accuracy, and today this constitutes a key component in genomic screens. Here, we cover the basic principles of RNA folding and touch upon some of the concepts in current methods that have been applied in genomic screens for de novo RNA structures in searches for novel ncRNA genes and regulatory RNA structure on mRNAs. We discuss the strengths and weaknesses of the different strategies and how they can complement each other.
非编码 RNA(ncRNA)不仅作为一类丰富的基因受到越来越多的关注,而且作为调节结构元件(一些位于 mRNA 中)。RNA 功能的一个关键特征是其结构。早期开发了计算方法来折叠和预测 RNA 结构,旨在辅助功能分析。随着越来越多的 ncRNA 的发现,很明显,这些 RNA 的很大一部分具有高度的结构。有趣的是,结构的很大一部分由规则的 Watson-Crick 和 GU 摆动碱基对组成。这一点以及可用基因组数量的增加,使得可以使用基于结构的方法进行基因组筛选。该领域已经从单个序列的折叠预测发展到使用 RNA 结构作为主要特征特征的基因组序列中的 ncRNA 计算筛选。早期的方法侧重于单个序列的能量导向折叠,而基于碱基对结构保持变化的比较分析在提高准确性方面非常有效,如今这构成了基因组筛选的关键组成部分。在这里,我们介绍了 RNA 折叠的基本原理,并讨论了当前方法中的一些概念,这些概念已应用于基因组筛选中,以寻找新的 ncRNA 基因和 mRNA 上的调节 RNA 结构。我们讨论了不同策略的优缺点以及它们如何相互补充。