Department of Biomedical Engineering, Johns Hopkins University, Baltimore, Maryland, United States of America.
PLoS Genet. 2011 Jul;7(7):e1002177. doi: 10.1371/journal.pgen.1002177. Epub 2011 Jul 28.
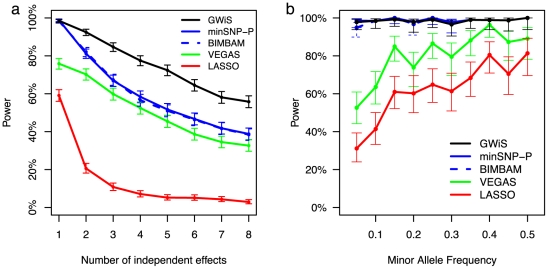
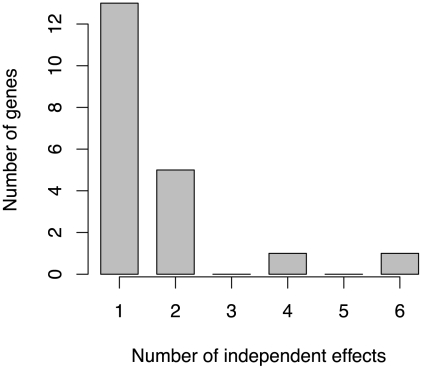
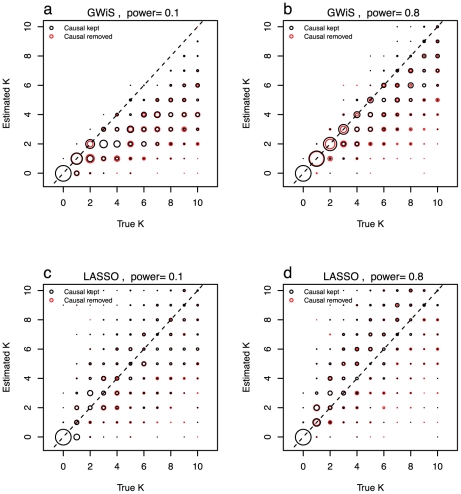
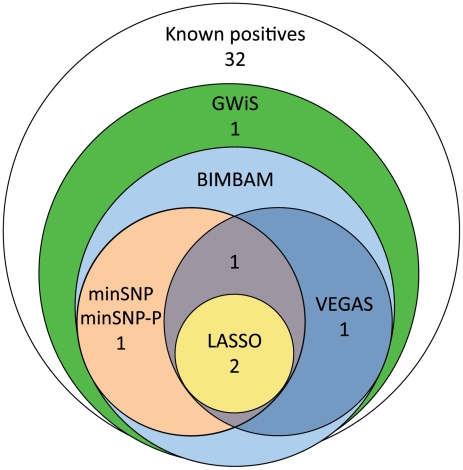
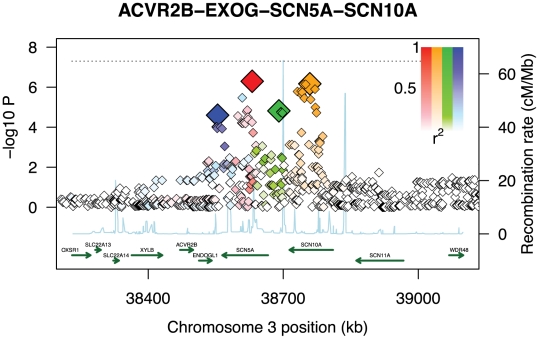
Genome-wide association studies (GWAS) are now used routinely to identify SNPs associated with complex human phenotypes. In several cases, multiple variants within a gene contribute independently to disease risk. Here we introduce a novel Gene-Wide Significance (GWiS) test that uses greedy Bayesian model selection to identify the independent effects within a gene, which are combined to generate a stronger statistical signal. Permutation tests provide p-values that correct for the number of independent tests genome-wide and within each genetic locus. When applied to a dataset comprising 2.5 million SNPs in up to 8,000 individuals measured for various electrocardiography (ECG) parameters, this method identifies more validated associations than conventional GWAS approaches. The method also provides, for the first time, systematic assessments of the number of independent effects within a gene and the fraction of disease-associated genes housing multiple independent effects, observed at 35%-50% of loci in our study. This method can be generalized to other study designs, retains power for low-frequency alleles, and provides gene-based p-values that are directly compatible for pathway-based meta-analysis.
全基因组关联研究(GWAS)现在被常规用于鉴定与复杂人类表型相关的 SNP。在某些情况下,基因内的多个变异独立地贡献于疾病风险。在这里,我们引入了一种新的基因范围显著性(GWiS)检验方法,该方法使用贪婪贝叶斯模型选择来识别基因内的独立效应,这些效应被组合以产生更强的统计信号。置换检验提供了 p 值,这些 p 值校正了全基因组和每个遗传位点上的独立检验数量。当应用于包含 250 万个 SNP 的数据集,这些 SNP 测量了 8000 多名个体的各种心电图(ECG)参数,与传统的 GWAS 方法相比,该方法识别出了更多经证实的关联。该方法还首次提供了对基因内独立效应数量和包含多个独立效应的疾病相关基因比例的系统评估,在我们的研究中,35%-50%的位点观察到了这些效应。该方法可以推广到其他研究设计,保留低频等位基因的功效,并提供基于基因的 p 值,这些 p 值可直接用于基于通路的荟萃分析。